
Hello Lise, This is in your own local instance? If so, the instance will need to have a few configuration changes and reference genomes added (and the server you are using must have the proper resources available to run the tools - the general rule is if you can run the tool line command, then it will almost certainly run in Galaxy - see the source binary for required resource information). If you are using a cloud Galaxy, the genomes can just be added. Resources on a cloud instance can be scaled up if you find that you need more (memory, more nodes, etc) to achieve the throughput you want, or to run large datasets. For a local instance, the basic install instructions are here: https://wiki.galaxyproject.org/Admin/GetGalaxy Then, you need to proceed to the Advanced Production configuration (#3) to here: https://wiki.galaxyproject.org/Admin/GetGalaxy#Advanced_Configuration Once you are ready to add genomes (local or cloud), make sure your tool dependencies are set up (Bowtie2, etc). The builds.txt file needs to list the genomes you are working with, then the genomes and indexes need to be added. We provide our genomes via rsync if you want to use them, or you can download a genome from any source you want and create indexes yourself. If you use our reference genomes, then it will be easier to use your data on the public server, if that is a future goal (using the same reference genome throughout an analysis is very important). Bowtie2 indexes are not yet available for all genomes, but these not difficult to create. Instructions for this are in these two wikis: https://wiki.galaxyproject.org/Admin/Data%20Integration https://wiki.galaxyproject.org/Admin/NGS%20Local%20Setup Hopefully this helps. If anything has been misunderstood, please let us know and provide more details. Please keep all replies on the mailing list so that the development community can help with troubleshooting. Best, Jen Galaxy team On 1/6/14 8:46 AM, Ilse van de Vondervoort wrote:
Dear developers,
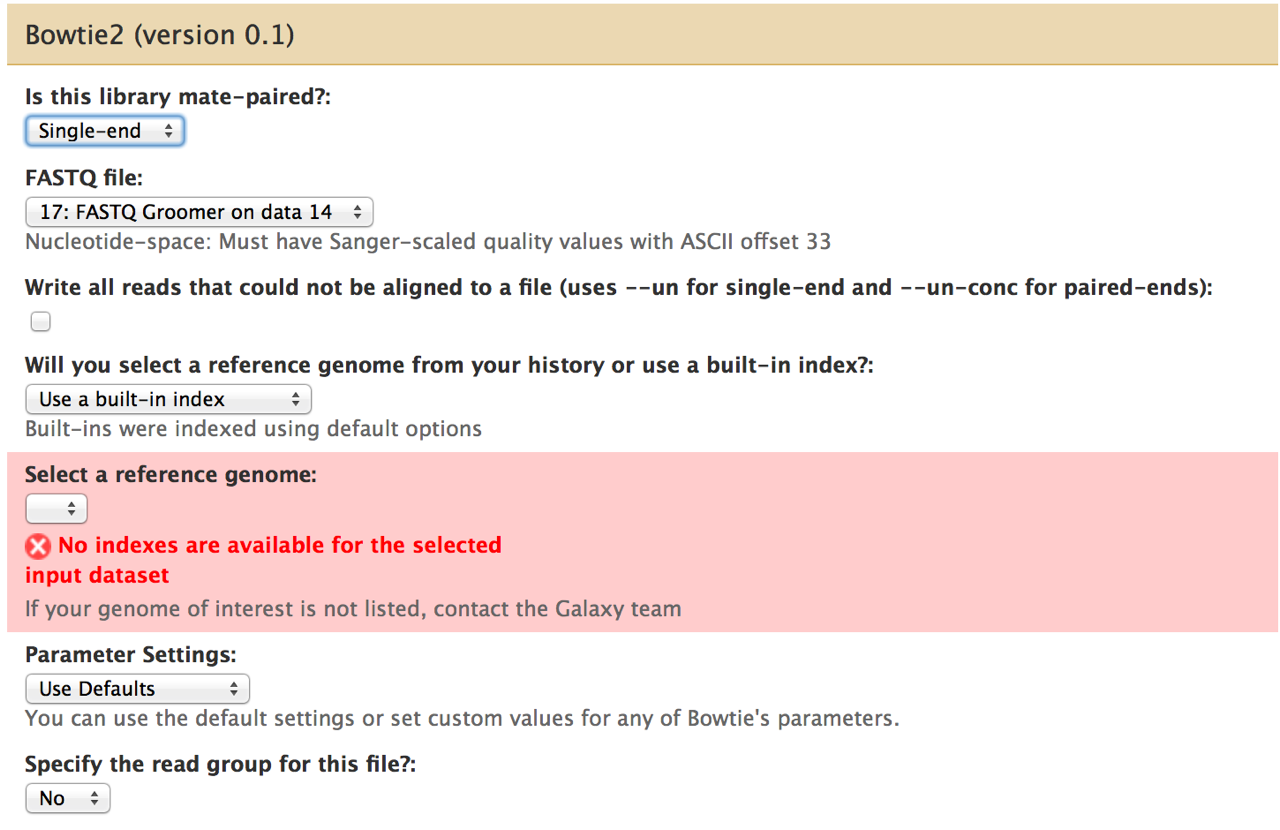
I encountered a problem when I wanted to map rat microRNA sequencing data (FASTQ file, after FASTQ Groomer) to the reference genome (see attached screenshot). I am unable to select any build-in reference genome, and I haven't used any myself thus I can't select one from my history. Hopefully, this is an easy to overcome issue.
Kind regards, Ilse
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
-- Jennifer Hillman-Jackson http://galaxyproject.org
{kind=link}