Concept for a Galaxy Versioned Fasta Data Retrieval Tool

We are about to implement a fasta database (file) versioning system as a Galaxy tool. I wanted to get interested people's feedback first before we roll ahead with the prototype implementation. The versioning system aims to: * Enable reproducible research: To recreate a search result at a certain point in time we need versioning so that search and mapping tools can look at sequence reference databases corresponding to a particular past date. This recall can also explain the difference between what was known in the past vs. currently. * Reduce hard drive space. Some databases are too big to keep N copies around, e.g. 5 years of 16S, updated monthly, is say, 670Mb + 668Mb + 665Mb + .... But occasionally we want to access past archives fairly quickly. * Integrate database versioning into Galaxy without adding a lot of complexity. A bonus would be to enable the efficient sharing of version databases between computers/servers. The solution we think would work centres around a "Versioned Data Retrieval" tool (draft image attached) that would work as follows: 1) User selects from a list of databases provided by "Shared Data > Data Libraries > Versioned Data". - Each database has a master file that keeps its various versions as a list of time-stamped insert/delete transactions of key (fasta id) value (description & sequence) pairs. - Each master file is managed outside of galaxy via a triggered process on regular fasta file imports from data sources like NCBI or other niche sources. - We're expecting, due to the nature of fasta archived sequence updates, that our master file would only be about 1.1x the latest version in size (uncompressed). 2) User enters date / version id to retrieve (validated) 3) If a cached version of that database exists, it is linked into user's history. 4) Otherwise a new version of it is created, placed in cache, and linked into history. - The cached version itself then shows up as linked data under a Data Library > Versioned Data subfolder. 5) User can select preconfigured workflow(s) to execute on the selected retreived fasta file to regenerate any database products they need. - Workflow output data would also be cached in the same way the fasta data is - by linking the Galaxy Data Library to it. - Workflow execution will be skipped if end data already exists in cache. - Simple makeblastdb or bowtie-build commands, or more specific workflows that include dustmasker etc can be implemented. Does this sound attractive? We're hoping such a vision could handle Fasta databases from 12mb to e.g. 200Gb (probably requires makeblastdb in parallel at that scale). Preliminary work suggests this project is doable via the Galaxy API without galaxy customization - does that sound right?! Feedback really appreciated! Regards, Damion Dooley Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada
{kind=link}

Hi Damion, the idea sounds fantastic! Can we go a step further and use a specific datatype that keeps entire fasta files versioned and the user can choose which version he wants to use, in any tool? Please have a look at my talk at GCC2012. Maybe you are interested in the (old) patches. I would be very interested to restart this old project. https://wiki.galaxyproject.org/Events/GCC2012/Abstracts#Keeping_Track_of_Lif... Am 23.08.2014 um 03:24 schrieb Dooley, Damion:
We are about to implement a fasta database (file) versioning system as a Galaxy tool. I wanted to get interested people's feedback first before we roll ahead with the prototype implementation. The versioning system aims to:
* Enable reproducible research: To recreate a search result at a certain point in time we need versioning so that search and mapping tools can look at sequence reference databases corresponding to a particular past date. This recall can also explain the difference between what was known in the past vs. currently.
* Reduce hard drive space. Some databases are too big to keep N copies around, e.g. 5 years of 16S, updated monthly, is say, 670Mb + 668Mb + 665Mb + .... But occasionally we want to access past archives fairly quickly.
* Integrate database versioning into Galaxy without adding a lot of complexity.
A bonus would be to enable the efficient sharing of version databases between computers/servers.
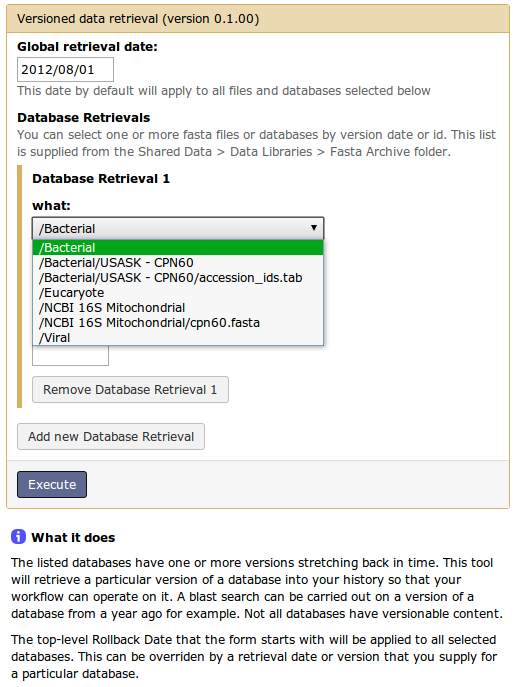
The solution we think would work centres around a "Versioned Data Retrieval" tool (draft image attached) that would work as follows:
1) User selects from a list of databases provided by "Shared Data > Data Libraries > Versioned Data". - Each database has a master file that keeps its various versions as a list of time-stamped insert/delete transactions of key (fasta id) value (description & sequence) pairs. - Each master file is managed outside of galaxy via a triggered process on regular fasta file imports from data sources like NCBI or other niche sources. - We're expecting, due to the nature of fasta archived sequence updates, that our master file would only be about 1.1x the latest version in size (uncompressed). 2) User enters date / version id to retrieve (validated) 3) If a cached version of that database exists, it is linked into user's history. 4) Otherwise a new version of it is created, placed in cache, and linked into history. - The cached version itself then shows up as linked data under a Data Library > Versioned Data subfolder. 5) User can select preconfigured workflow(s) to execute on the selected retreived fasta file to regenerate any database products they need. - Workflow output data would also be cached in the same way the fasta data is - by linking the Galaxy Data Library to it. - Workflow execution will be skipped if end data already exists in cache. - Simple makeblastdb or bowtie-build commands, or more specific workflows that include dustmasker etc can be implemented.
Does this sound attractive?
I think all of the use cases are covered by the old project mentioned above. But I did not create a new tool I have created a new 'select type' everyone can use in all tools. It was using git underneath (yeah, I have the entire PDB in git and it is working fine :)) but we can probably change git with a database if you like. To answer your question: Yes, very attractive!
We're hoping such a vision could handle Fasta databases from 12mb to e.g. 200Gb (probably requires makeblastdb in parallel at that scale).
Preliminary work suggests this project is doable via the Galaxy API without galaxy customization - does that sound right?!
Yes, as long as the User has an API key. Cheers, Bjoern
Feedback really appreciated!
Regards,
Damion Dooley
Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
Ok, I'll be very happy to see what you've accomplished there. I will read through what you've done when I return from vacation in a week! A key need is to have whatever data comes in show up as linked data in one's history to avoid server overhead; a second objective was to not need to modify existing workflows - as long as they could work of data in history that is typed appropriately. So your 'select type' solution sounds intreguing! And certainly interested in your use of git - I tried using git, using a 1-line fasta data format, but git seemed to choke on protein fasta files? And did it run into performance problems with larger files? That was my experience. I think I read its authors say that its upper limit was 15gb. That was the motivation for writing a simple key-value master file diff system that seems to have the same I/O as git on smaller files, but more reliable for the fasta data case, and no problems with larger files - it outputs a new version in the same time it takes to read a master file. It has drawbacks though - incoming data to compare master with must be sorted in 1 line fasta format first. Thanks for your input; looking forward to your project writeup... Damion Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada ________________________________________ From: Björn Grüning [bjoern.gruening@gmail.com] Sent: Saturday, August 23, 2014 12:17 AM To: Dooley, Damion; galaxy-dev@lists.bx.psu.edu Cc: Hsiao, William Subject: Re: [galaxy-dev] Concept for a Galaxy Versioned Fasta Data Retrieval Tool Hi Damion, the idea sounds fantastic! Can we go a step further and use a specific datatype that keeps entire fasta files versioned and the user can choose which version he wants to use, in any tool? Please have a look at my talk at GCC2012. Maybe you are interested in the (old) patches. I would be very interested to restart this old project. https://wiki.galaxyproject.org/Events/GCC2012/Abstracts#Keeping_Track_of_Lif... Am 23.08.2014 um 03:24 schrieb Dooley, Damion:
We are about to implement a fasta database (file) versioning system as a Galaxy tool. I wanted to get interested people's feedback first before we roll ahead with the prototype implementation. The versioning system aims to: .... - Simple makeblastdb or bowtie-build commands, or more specific workflows that include dustmasker etc can be implemented.
Does this sound attractive?
I think all of the use cases are covered by the old project mentioned above. But I did not create a new tool I have created a new 'select type' everyone can use in all tools. It was using git underneath (yeah, I have the entire PDB in git and it is working fine :)) but we can probably change git with a database if you like. To answer your question: Yes, very attractive!
We're hoping such a vision could handle Fasta databases from 12mb to e.g. 200Gb (probably requires makeblastdb in parallel at that scale).
Preliminary work suggests this project is doable via the Galaxy API without galaxy customization - does that sound right?!
Yes, as long as the User has an API key. Cheers, Bjoern

Damion, Thanks a lot - consequently treating the toppic of 'reproducable science' is a competition, but absolutely required. Björn really touched my mind when he gave the linked talk in Chicago (GCC 2012). Although for a longer time things got stuck, I think that Galaxy is still a (the?) key to it, because the principal structures allow it - it's somehow native to it. Since some important and powerful elements came up (think of e.g. the API), the gap for reaching reproducability also from the reference side using 'on-board tools' of the framework (without bending or disassembling the code too strong) has hardly narrowed. Due to the medical context our instance is in, we really need features like this. In some particular detail I would maybe implement the respective functionalities a bit different (due to performance), but in vast majority I agree: this sounds attractive! Marius' remark on data managers (which are brand new as far as I understood the GCC talks) sounds reasonable, although I did not get in touch with it yet. So, count me in, I'm already a bit excited :). Cheers, Sebastian Dooley, Damion schrieb:
Ok, I'll be very happy to see what you've accomplished there. I will read through what you've done when I return from vacation in a week!
A key need is to have whatever data comes in show up as linked data in one's history to avoid server overhead; a second objective was to not need to modify existing workflows - as long as they could work of data in history that is typed appropriately. So your 'select type' solution sounds intreguing!
And certainly interested in your use of git - I tried using git, using a 1-line fasta data format, but git seemed to choke on protein fasta files? And did it run into performance problems with larger files? That was my experience. I think I read its authors say that its upper limit was 15gb. That was the motivation for writing a simple key-value master file diff system that seems to have the same I/O as git on smaller files, but more reliable for the fasta data case, and no problems with larger files - it outputs a new version in the same time it takes to read a master file. It has drawbacks though - incoming data to compare master with must be sorted in 1 line fasta format first.
Thanks for your input; looking forward to your project writeup...
Damion
Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada ________________________________________ From: Björn Grüning [bjoern.gruening@gmail.com] Sent: Saturday, August 23, 2014 12:17 AM To: Dooley, Damion; galaxy-dev@lists.bx.psu.edu Cc: Hsiao, William Subject: Re: [galaxy-dev] Concept for a Galaxy Versioned Fasta Data Retrieval Tool
Hi Damion,
the idea sounds fantastic! Can we go a step further and use a specific datatype that keeps entire fasta files versioned and the user can choose which version he wants to use, in any tool? Please have a look at my talk at GCC2012. Maybe you are interested in the (old) patches. I would be very interested to restart this old project.
https://wiki.galaxyproject.org/Events/GCC2012/Abstracts#Keeping_Track_of_Lif...
Am 23.08.2014 um 03:24 schrieb Dooley, Damion:
We are about to implement a fasta database (file) versioning system as a Galaxy tool. I wanted to get interested people's feedback first before we roll ahead with the prototype implementation. The versioning system aims to: .... - Simple makeblastdb or bowtie-build commands, or more specific workflows that include dustmasker etc can be implemented.
Does this sound attractive? I think all of the use cases are covered by the old project mentioned above. But I did not create a new tool I have created a new 'select type' everyone can use in all tools. It was using git underneath (yeah, I have the entire PDB in git and it is working fine :)) but we can probably change git with a database if you like.
To answer your question: Yes, very attractive!
We're hoping such a vision could handle Fasta databases from 12mb to e.g. 200Gb (probably requires makeblastdb in parallel at that scale).
Preliminary work suggests this project is doable via the Galaxy API without galaxy customization - does that sound right?! Yes, as long as the User has an API key.
Cheers, Bjoern ___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
-- Sebastian Schaaf, M.Sc. Bioinformatics Faculty Coordinator NGS Infrastructure Chair of Biometry and Bioinformatics Department of Medical Informatics, Biometry and Epidemiology (IBE) University of Munich Marchioninistr. 15, K U1 (postal) Marchioninistr. 17, U 006 (office) D-81377 Munich (Germany) Tel: +49 89 2180-78178
Am 25.08.2014 um 18:05 schrieb Dooley, Damion:
Ok, I'll be very happy to see what you've accomplished there. I will read through what you've done when I return from vacation in a week!
A key need is to have whatever data comes in show up as linked data in one's history to avoid server overhead; a second objective was to not need to modify existing workflows - as long as they could work of data in history that is typed appropriately. So your 'select type' solution sounds intreguing!
And certainly interested in your use of git - I tried using git, using a 1-line fasta data format, but git seemed to choke on protein fasta files? And did it run into performance problems with larger files? That was my experience. I think I read its authors say that its upper limit was 15gb.
This is probably true for one large file. I'm storing the entire PDB in git since a few years. One entry one file and it works fine. Do you know git annex? https://git-annex.branchable.com/
That was the motivation for writing a simple key-value master file diff system that seems to have the same I/O as git on smaller files, but more reliable for the fasta data case, and no problems with larger files - it outputs a new version in the same time it takes to read a master file. It has drawbacks though - incoming data to compare master with must be sorted in 1 line fasta format first.
My intention was to create a universal solution for database tracking. So if you can please design your system in such a way that you can store arbitrary data, not only fasta files.
Thanks for your input; looking forward to your project writeup...
Wonderful! Talk to you after your holidays! Bjoern
Damion
Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada ________________________________________ From: Björn Grüning [bjoern.gruening@gmail.com] Sent: Saturday, August 23, 2014 12:17 AM To: Dooley, Damion; galaxy-dev@lists.bx.psu.edu Cc: Hsiao, William Subject: Re: [galaxy-dev] Concept for a Galaxy Versioned Fasta Data Retrieval Tool
Hi Damion,
the idea sounds fantastic! Can we go a step further and use a specific datatype that keeps entire fasta files versioned and the user can choose which version he wants to use, in any tool? Please have a look at my talk at GCC2012. Maybe you are interested in the (old) patches. I would be very interested to restart this old project.
https://wiki.galaxyproject.org/Events/GCC2012/Abstracts#Keeping_Track_of_Lif...
Am 23.08.2014 um 03:24 schrieb Dooley, Damion:
We are about to implement a fasta database (file) versioning system as a Galaxy tool. I wanted to get interested people's feedback first before we roll ahead with the prototype implementation. The versioning system aims to: .... - Simple makeblastdb or bowtie-build commands, or more specific workflows that include dustmasker etc can be implemented.
Does this sound attractive?
I think all of the use cases are covered by the old project mentioned above. But I did not create a new tool I have created a new 'select type' everyone can use in all tools. It was using git underneath (yeah, I have the entire PDB in git and it is working fine :)) but we can probably change git with a database if you like.
To answer your question: Yes, very attractive!
We're hoping such a vision could handle Fasta databases from 12mb to e.g. 200Gb (probably requires makeblastdb in parallel at that scale).
Preliminary work suggests this project is doable via the Galaxy API without galaxy customization - does that sound right?!
Yes, as long as the User has an API key.
Cheers, Bjoern
Hi, There have been a few comments about how general we could make the system for Galaxy use or just as a stand-alone command line driven tool. So some notes below about what I could see it taking on. Given the scale of the sequencing data problem, I'm sure the Galaxy community has important feedback on this. I looked at git annex and it appears to me that though it promises to keep track of and synchronize network located files, it doesn't do versioning on them - am I wrong about that? I also looked at https://code.google.com/p/leveldb/ , also a key value database which relies more heavily on indexes - but I see that though this is well-tuned to answering key queries, it isn't particularly good at storing and retrieving entire versions of a database that could be many gigabytes long, which is our mission. It is relatively easy to generalize the simple keydb prototype I wrote so that it can handle any key-value database - including binary content and even binary key data, not just text (fasta sequences). So a name change for the tool is a good idea. I want a versioning system that doesn't assume the incoming master file of key-value pairs is in the same order as it was on a previous import run. I was afraid that any arbitrary change in the order of content on the source server could completely destroy the efficiency of a differential approach. Git assumes its content is like a document - so it generates a slew of inserts and deletes, in fact provides no benefit, if the fasta entries are rearranged. I tested helping git overcome this hurdle by converting the fasta content to 1 line key/value fasta entries, and sorting them before git processing. That seemed to work for some smaller and larger nucleotide fasta files (tested 10m to 2gb) but failed when it came to processing protein fasta files; though possibly that was because of the fasta data line length. That became another concern - thinking that git was failing because each line of the input file was many thousands of characters long. So having done a "keydb" versioning engine that works and performs as well as git, I am definitely shying away from git now as unreliable on certain kinds of data. The keydb approach is able to generate a version file at about the same speed that it takes to read the latest version of the same db, i.e. at 50mb/s on a standard hard drive. An extension to keydb that enables it to take in just a list of adds or deletes or updates is desirable but that can come later. More efficiency can be had by fine-tuning the updates so that one whole line of key-value doesn't have to replace the previous one but that's for later too. A generalization note that the keydb approach works where the keys are a sparse array. There's nothing stopping the keys from representing a 2D or 3D sparse array of data as long as the coordinates are coded uniquely into the one key list. For those interested in versioning XML data there is an interesting summary of the challenges here: http://useless-factor.blogspot.ca/2008/01/matching-diffing-and-merging-xml.h... . It leaves me thinking that quick versioning of xml data could only be accomplished if it could somehow be converted into a key-value db, i.e. with each top level xml record identified by a unique key. I could see breaking larger keydb databases up into smaller chunks for data retrieval and fast parallel processing - the usual approach being to separate the sorted key-value db out into files based on the first character or two in the key of each record. Does this go along with people's expectations? Cheers, Damion ________________________________________ From: Björn Grüning [bjoern.gruening@gmail.com] Sent: Monday, September 01, 2014 12:47 PM To: Dooley, Damion; Björn Grüning; galaxy-dev@lists.bx.psu.edu Cc: Hsiao, William Subject: Re: [galaxy-dev] Concept for a Galaxy Versioned Fasta Data Retrieval Tool Am 25.08.2014 um 18:05 schrieb Dooley, Damion:
Ok, I'll be very happy to see what you've accomplished there. I will read through what you've done when I return from vacation in a week!
A key need is to have whatever data comes in show up as linked data in one's history to avoid server overhead; a second objective was to not need to modify existing workflows - as long as they could work of data in history that is typed appropriately. So your 'select type' solution sounds intreguing!
And certainly interested in your use of git - I tried using git, using a 1-line fasta data format, but git seemed to choke on protein fasta files? And did it run into performance problems with larger files? That was my experience. I think I read its authors say that its upper limit was 15gb.
This is probably true for one large file. I'm storing the entire PDB in git since a few years. One entry one file and it works fine. Do you know git annex? https://git-annex.branchable.com/
That was the motivation for writing a simple key-value master file diff system that seems to have the same I/O as git on smaller files, but more reliable for the fasta data case, and no problems with larger files - it outputs a new version in the same time it takes to read a master file. It has drawbacks though - incoming data to compare master with must be sorted in 1 line fasta format first.
My intention was to create a universal solution for database tracking. So if you can please design your system in such a way that you can store arbitrary data, not only fasta files.

I also think that this a great idea, and as you described it I think it's feasible as a stand-alone galaxy tool. Eventually you consider to implement this as a data manager ( https://wiki.galaxyproject.org/Admin/Tools/DataManagers) ? On 23 August 2014 03:24, Dooley, Damion <Damion.Dooley@bccdc.ca> wrote:
We are about to implement a fasta database (file) versioning system as a Galaxy tool. I wanted to get interested people's feedback first before we roll ahead with the prototype implementation. The versioning system aims to:
* Enable reproducible research: To recreate a search result at a certain point in time we need versioning so that search and mapping tools can look at sequence reference databases corresponding to a particular past date. This recall can also explain the difference between what was known in the past vs. currently.
* Reduce hard drive space. Some databases are too big to keep N copies around, e.g. 5 years of 16S, updated monthly, is say, 670Mb + 668Mb + 665Mb + .... But occasionally we want to access past archives fairly quickly.
* Integrate database versioning into Galaxy without adding a lot of complexity.
A bonus would be to enable the efficient sharing of version databases between computers/servers.
The solution we think would work centres around a "Versioned Data Retrieval" tool (draft image attached) that would work as follows:
1) User selects from a list of databases provided by "Shared Data > Data Libraries > Versioned Data". - Each database has a master file that keeps its various versions as a list of time-stamped insert/delete transactions of key (fasta id) value (description & sequence) pairs. - Each master file is managed outside of galaxy via a triggered process on regular fasta file imports from data sources like NCBI or other niche sources. - We're expecting, due to the nature of fasta archived sequence updates, that our master file would only be about 1.1x the latest version in size (uncompressed). 2) User enters date / version id to retrieve (validated) 3) If a cached version of that database exists, it is linked into user's history. 4) Otherwise a new version of it is created, placed in cache, and linked into history. - The cached version itself then shows up as linked data under a Data Library > Versioned Data subfolder. 5) User can select preconfigured workflow(s) to execute on the selected retreived fasta file to regenerate any database products they need. - Workflow output data would also be cached in the same way the fasta data is - by linking the Galaxy Data Library to it. - Workflow execution will be skipped if end data already exists in cache. - Simple makeblastdb or bowtie-build commands, or more specific workflows that include dustmasker etc can be implemented.
Does this sound attractive?
We're hoping such a vision could handle Fasta databases from 12mb to e.g. 200Gb (probably requires makeblastdb in parallel at that scale).
Preliminary work suggests this project is doable via the Galaxy API without galaxy customization - does that sound right?!
Feedback really appreciated!
Regards,
Damion Dooley
Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada ___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
I'll have a good look at the data manager approach - a quick glance tells me it is designed to fetch data and so be able to get fresh data on demand. We'll need a 3rd party data import process that operates on its own pre-defined schedule so that each data file gets to have its own update schedule - weekly/monthly/yearly. So it seems the galaxy API could accomplish this in talking with the data manager system? I was going to look into the http://biomaj.genouest.org/ system as another possibility. But is the data manager approach able to get data in such a way that it can just place it into a user's history as a linked file or set of files? I.e. the solution we need has its master databases locally on the server, and will generate a cached version from the master file - without any networking off of the server. I'm back from vacation in a week so I'll get to check that out in more detail... Cheers, Damion Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada ________________________________________ From: Marius van den Beek [m.vandenbeek@gmail.com] Sent: Saturday, August 23, 2014 2:25 AM To: Dooley, Damion Cc: galaxy-dev@lists.bx.psu.edu; Hsiao, William Subject: Re: [galaxy-dev] Concept for a Galaxy Versioned Fasta Data Retrieval Tool I also think that this a great idea, and as you described it I think it's feasible as a stand-alone galaxy tool. Eventually you consider to implement this as a data manager (https://wiki.galaxyproject.org/Admin/Tools/DataManagers) ?

I would ask that such a tool be designed to work well in a stand-alone mode outside of Galaxy. Cheers, On Sat, Aug 23, 2014 at 4:24 AM, Dooley, Damion <Damion.Dooley@bccdc.ca> wrote:
We are about to implement a fasta database (file) versioning system as a Galaxy tool. I wanted to get interested people's feedback first before we roll ahead with the prototype implementation. The versioning system aims to:
* Enable reproducible research: To recreate a search result at a certain point in time we need versioning so that search and mapping tools can look at sequence reference databases corresponding to a particular past date. This recall can also explain the difference between what was known in the past vs. currently.
* Reduce hard drive space. Some databases are too big to keep N copies around, e.g. 5 years of 16S, updated monthly, is say, 670Mb + 668Mb + 665Mb + .... But occasionally we want to access past archives fairly quickly.
* Integrate database versioning into Galaxy without adding a lot of complexity.
A bonus would be to enable the efficient sharing of version databases between computers/servers.
The solution we think would work centres around a "Versioned Data Retrieval" tool (draft image attached) that would work as follows:
1) User selects from a list of databases provided by "Shared Data > Data Libraries > Versioned Data". - Each database has a master file that keeps its various versions as a list of time-stamped insert/delete transactions of key (fasta id) value (description & sequence) pairs. - Each master file is managed outside of galaxy via a triggered process on regular fasta file imports from data sources like NCBI or other niche sources. - We're expecting, due to the nature of fasta archived sequence updates, that our master file would only be about 1.1x the latest version in size (uncompressed). 2) User enters date / version id to retrieve (validated) 3) If a cached version of that database exists, it is linked into user's history. 4) Otherwise a new version of it is created, placed in cache, and linked into history. - The cached version itself then shows up as linked data under a Data Library > Versioned Data subfolder. 5) User can select preconfigured workflow(s) to execute on the selected retreived fasta file to regenerate any database products they need. - Workflow output data would also be cached in the same way the fasta data is - by linking the Galaxy Data Library to it. - Workflow execution will be skipped if end data already exists in cache. - Simple makeblastdb or bowtie-build commands, or more specific workflows that include dustmasker etc can be implemented.
Does this sound attractive?
We're hoping such a vision could handle Fasta databases from 12mb to e.g. 200Gb (probably requires makeblastdb in parallel at that scale).
Preliminary work suggests this project is doable via the Galaxy API without galaxy customization - does that sound right?!
Feedback really appreciated!
Regards,
Damion Dooley
Hsiao lab, BC Public Health Microbiology & Reference Laboratory, BC Centre for Disease Control 655 West 12th Avenue, Vancouver, British Columbia, V5Z 4R4 Canada ___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
-- Michael R. Crusoe: Programmer & Bioinformatician mcrusoe@msu.edu @ the Genomics, Evolution, and Development lab; Michigan State U http://ged.msu.edu/ http://orcid.org/0000-0002-2961-9670 @biocrusoe <http://twitter.com/biocrusoe>
participants (5)
-
 Björn Grüning
Björn Grüning -
 Dooley, Damion
Dooley, Damion -
 Marius van den Beek
Marius van den Beek -
 Michael R. Crusoe
Michael R. Crusoe -
 Sebastian Schaaf
Sebastian Schaaf