problems with 'action type="metadata" name="dbkey"' in tool xml

Dear All I am trying to move our Galaxy installation to new hardware and at the same time to a new version (the original is "15.10",minor version "2" via mercurial; the new is "16.04" via github): So far, I am making good progress, but I am struggling with a tool which was written by someone else who has left more than 2 years ago. The problem is: it doesn't set the dbkey correctly. I get: <Cheetah.Template.Template object at 0x7f1e40210f50> The tool xml is as follows: <inputs> <param name="sample_file" type="data" format="qsamples,qsamplespaired" label="Sample File" help="set of sequence files created by the 'select sequence files' tool"> </param> <param name="index" type="select" label="Reference Genome" help="all reads will be mapped to this reference"> <options from_data_table="fmi__quasr_genomes"/> </param> ... </inputs> <outputs> <data name="output01" type="data" format="qproject" label="qProject of ${sample_file.name}"> <actions> <action type="metadata" name="dbkey"> <option type="from_param" name="index"/> </action> </actions> </data> </outputs> The user can select a genome (based on the Bioconductor BSgenome packages) from the tab delimited fmi__quasr_genomes table, which looks like: hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) BSgenome.Hsapiens.UCSC.hg19 hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) BSgenome.Hsapiens.UCSC.hg18 ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) BSgenome.Celegans.UCSC.ce10 ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) BSgenome.Celegans.UCSC.ce6 ... The tool works: - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) - once the job is submitted, it uses the right Bioconductor package (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc) However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: <Cheetah.Template.Template object at 0x7f1e40210f50> Has anything changed with the 'action' tag since release 15.10? which makes this simple xml no longer working? I have to admit, it does look different than the example on the wiki (https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t... ). I tried the code from the wiki which is based on the bowtie_wrapper (https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...) as well: <actions> <action type="metadata" name="dbkey"> <option type="from_data_table" name="fmi__quasr_genomes" column="1" offset="0"> <filter type="param_value" column="0" value="#" compare="startswith" keep="False"/> <filter type="param_value" ref="index" column="0"/> </option> </action> </actions> but it results in the same kind of error: <Cheetah.Template.Template object at 0x7ff82c0e43d0> Thank you very much for any help/comments Regards, Hans-Rudolf -- Hans-Rudolf Hotz, PhD Bioinformatics Support Friedrich Miescher Institute for Biomedical Research Maulbeerstrasse 66 4058 Basel/Switzerland

Hi Hans, see my comment below. On 16/08/16 09:51, Hans-Rudolf Hotz wrote:
Dear All
I am trying to move our Galaxy installation to new hardware and at the same time to a new version (the original is "15.10",minor version "2" via mercurial; the new is "16.04" via github):
So far, I am making good progress, but I am struggling with a tool which was written by someone else who has left more than 2 years ago. The problem is: it doesn't set the dbkey correctly.
I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
The tool xml is as follows:
<inputs> <param name="sample_file" type="data" format="qsamples,qsamplespaired" label="Sample File" help="set of sequence files created by the 'select sequence files' tool"> </param> <param name="index" type="select" label="Reference Genome" help="all reads will be mapped to this reference"> <options from_data_table="fmi__quasr_genomes"/> </param>
... </inputs> <outputs> <data name="output01" type="data" format="qproject" label="qProject of ${sample_file.name}"> <actions> <action type="metadata" name="dbkey"> <option type="from_param" name="index"/>
It seems you are missing a param_attribute="dbkey" here, it should be: <option type="from_param" name="index" param_attribute="dbkey"/> Cheers, Nicola
</action> </actions> </data> </outputs>
The user can select a genome (based on the Bioconductor BSgenome packages) from the tab delimited fmi__quasr_genomes table, which looks like:
hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) BSgenome.Hsapiens.UCSC.hg19 hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) BSgenome.Hsapiens.UCSC.hg18 ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) BSgenome.Celegans.UCSC.ce10 ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) BSgenome.Celegans.UCSC.ce6 ...
The tool works: - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) - once the job is submitted, it uses the right Bioconductor package (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc)
However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
Has anything changed with the 'action' tag since release 15.10? which makes this simple xml no longer working?
I have to admit, it does look different than the example on the wiki (https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t... ). I tried the code from the wiki which is based on the bowtie_wrapper (https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...) as well:
<actions> <action type="metadata" name="dbkey"> <option type="from_data_table" name="fmi__quasr_genomes" column="1" offset="0"> <filter type="param_value" column="0" value="#" compare="startswith" keep="False"/> <filter type="param_value" ref="index" column="0"/> </option> </action> </actions>
but it results in the same kind of error: <Cheetah.Template.Template object at 0x7ff82c0e43d0>
Thank you very much for any help/comments
Regards, Hans-Rudolf
Forget about my previous comment, I should have read your email better, sorry! Your code similar to bowtie_wrapper is also used by https://github.com/galaxyproject/tools-iuc/blob/master/tools/rgrnastar/rg_rn... and I think it should work, not sure if it's a regression in Galaxy. Cheers, Nicola On 16/08/16 11:29, Nicola Soranzo wrote:
Hi Hans, see my comment below.
On 16/08/16 09:51, Hans-Rudolf Hotz wrote:
Dear All
I am trying to move our Galaxy installation to new hardware and at the same time to a new version (the original is "15.10",minor version "2" via mercurial; the new is "16.04" via github):
So far, I am making good progress, but I am struggling with a tool which was written by someone else who has left more than 2 years ago. The problem is: it doesn't set the dbkey correctly.
I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
The tool xml is as follows:
<inputs> <param name="sample_file" type="data" format="qsamples,qsamplespaired" label="Sample File" help="set of sequence files created by the 'select sequence files' tool"> </param> <param name="index" type="select" label="Reference Genome" help="all reads will be mapped to this reference"> <options from_data_table="fmi__quasr_genomes"/> </param>
... </inputs> <outputs> <data name="output01" type="data" format="qproject" label="qProject of ${sample_file.name}"> <actions> <action type="metadata" name="dbkey"> <option type="from_param" name="index"/>
It seems you are missing a param_attribute="dbkey" here, it should be: <option type="from_param" name="index" param_attribute="dbkey"/>
Cheers, Nicola
</action> </actions> </data> </outputs>
The user can select a genome (based on the Bioconductor BSgenome packages) from the tab delimited fmi__quasr_genomes table, which looks like:
hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) BSgenome.Hsapiens.UCSC.hg19 hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) BSgenome.Hsapiens.UCSC.hg18 ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) BSgenome.Celegans.UCSC.ce10 ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) BSgenome.Celegans.UCSC.ce6 ...
The tool works: - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) - once the job is submitted, it uses the right Bioconductor package (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc)
However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
Has anything changed with the 'action' tag since release 15.10? which makes this simple xml no longer working?
I have to admit, it does look different than the example on the wiki (https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t... ). I tried the code from the wiki which is based on the bowtie_wrapper (https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...) as well:
<actions> <action type="metadata" name="dbkey"> <option type="from_data_table" name="fmi__quasr_genomes" column="1" offset="0"> <filter type="param_value" column="0" value="#" compare="startswith" keep="False"/> <filter type="param_value" ref="index" column="0"/> </option> </action> </actions>
but it results in the same kind of error: <Cheetah.Template.Template object at 0x7ff82c0e43d0>
Thank you very much for any help/comments
Regards, Hans-Rudolf
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
I've tested bowtie_wrapper on Galaxy release_16.04: <option type="from_data_table" name="bowtie_indexes" column="1" offset="0"> works fine for me. Cheers, Nicola On 16/08/16 11:42, Nicola Soranzo wrote:
Forget about my previous comment, I should have read your email better, sorry!
Your code similar to bowtie_wrapper is also used by https://github.com/galaxyproject/tools-iuc/blob/master/tools/rgrnastar/rg_rn... and I think it should work, not sure if it's a regression in Galaxy.
Cheers, Nicola
On 16/08/16 11:29, Nicola Soranzo wrote:
Hi Hans, see my comment below.
On 16/08/16 09:51, Hans-Rudolf Hotz wrote:
Dear All
I am trying to move our Galaxy installation to new hardware and at the same time to a new version (the original is "15.10",minor version "2" via mercurial; the new is "16.04" via github):
So far, I am making good progress, but I am struggling with a tool which was written by someone else who has left more than 2 years ago. The problem is: it doesn't set the dbkey correctly.
I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
The tool xml is as follows:
<inputs> <param name="sample_file" type="data" format="qsamples,qsamplespaired" label="Sample File" help="set of sequence files created by the 'select sequence files' tool"> </param> <param name="index" type="select" label="Reference Genome" help="all reads will be mapped to this reference"> <options from_data_table="fmi__quasr_genomes"/> </param>
... </inputs> <outputs> <data name="output01" type="data" format="qproject" label="qProject of ${sample_file.name}"> <actions> <action type="metadata" name="dbkey"> <option type="from_param" name="index"/>
It seems you are missing a param_attribute="dbkey" here, it should be: <option type="from_param" name="index" param_attribute="dbkey"/>
Cheers, Nicola
</action> </actions> </data> </outputs>
The user can select a genome (based on the Bioconductor BSgenome packages) from the tab delimited fmi__quasr_genomes table, which looks like:
hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) BSgenome.Hsapiens.UCSC.hg19 hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) BSgenome.Hsapiens.UCSC.hg18 ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) BSgenome.Celegans.UCSC.ce10 ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) BSgenome.Celegans.UCSC.ce6 ...
The tool works: - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) - once the job is submitted, it uses the right Bioconductor package (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc)
However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
Has anything changed with the 'action' tag since release 15.10? which makes this simple xml no longer working?
I have to admit, it does look different than the example on the wiki (https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t... ). I tried the code from the wiki which is based on the bowtie_wrapper (https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...) as well:
<actions> <action type="metadata" name="dbkey"> <option type="from_data_table" name="fmi__quasr_genomes" column="1" offset="0"> <filter type="param_value" column="0" value="#" compare="startswith" keep="False"/> <filter type="param_value" ref="index" column="0"/> </option> </action> </actions>
but it results in the same kind of error: <Cheetah.Template.Template object at 0x7ff82c0e43d0>
Thank you very much for any help/comments
Regards, Hans-Rudolf
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
Hi Nicola Many thanks for your comments/suggestions. I've just tested the rnaStar wrapper, and it works as well. I will try to dig deeper Hans-Rudolf On 08/16/2016 01:38 PM, Nicola Soranzo wrote:
I've tested bowtie_wrapper on Galaxy release_16.04: <option type="from_data_table" name="bowtie_indexes" column="1" offset="0"> works fine for me.
Cheers, Nicola
On 16/08/16 11:42, Nicola Soranzo wrote:
Forget about my previous comment, I should have read your email better, sorry!
Your code similar to bowtie_wrapper is also used by https://github.com/galaxyproject/tools-iuc/blob/master/tools/rgrnastar/rg_rn... and I think it should work, not sure if it's a regression in Galaxy.
Cheers, Nicola
On 16/08/16 11:29, Nicola Soranzo wrote:
Hi Hans, see my comment below.
On 16/08/16 09:51, Hans-Rudolf Hotz wrote:
Dear All
I am trying to move our Galaxy installation to new hardware and at the same time to a new version (the original is "15.10",minor version "2" via mercurial; the new is "16.04" via github):
So far, I am making good progress, but I am struggling with a tool which was written by someone else who has left more than 2 years ago. The problem is: it doesn't set the dbkey correctly.
I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
The tool xml is as follows:
<inputs> <param name="sample_file" type="data" format="qsamples,qsamplespaired" label="Sample File" help="set of sequence files created by the 'select sequence files' tool"> </param> <param name="index" type="select" label="Reference Genome" help="all reads will be mapped to this reference"> <options from_data_table="fmi__quasr_genomes"/> </param>
... </inputs> <outputs> <data name="output01" type="data" format="qproject" label="qProject of ${sample_file.name}"> <actions> <action type="metadata" name="dbkey"> <option type="from_param" name="index"/>
It seems you are missing a param_attribute="dbkey" here, it should be: <option type="from_param" name="index" param_attribute="dbkey"/>
Cheers, Nicola
</action> </actions> </data> </outputs>
The user can select a genome (based on the Bioconductor BSgenome packages) from the tab delimited fmi__quasr_genomes table, which looks like:
hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) BSgenome.Hsapiens.UCSC.hg19 hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) BSgenome.Hsapiens.UCSC.hg18 ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) BSgenome.Celegans.UCSC.ce10 ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) BSgenome.Celegans.UCSC.ce6 ...
The tool works: - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) - once the job is submitted, it uses the right Bioconductor package (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc)
However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
Has anything changed with the 'action' tag since release 15.10? which makes this simple xml no longer working?
I have to admit, it does look different than the example on the wiki (https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t... ). I tried the code from the wiki which is based on the bowtie_wrapper (https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...) as well:
<actions> <action type="metadata" name="dbkey"> <option type="from_data_table" name="fmi__quasr_genomes" column="1" offset="0"> <filter type="param_value" column="0" value="#" compare="startswith" keep="False"/> <filter type="param_value" ref="index" column="0"/> </option> </action> </actions>
but it results in the same kind of error: <Cheetah.Template.Template object at 0x7ff82c0e43d0>
Thank you very much for any help/comments
Regards, Hans-Rudolf
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
Hi Nicola and others on the list It looks like it has something to do with the 'conditional' I took the rnaStar xml file, and reduced it to the bare bones, just to reproduce the issue (see attachment: rg_rnaStar.simple ). This works. It picks up the right dbkey. I made the xml file even simpler and removed the 'conditional' (see attachment: rg_rnaStar.simple_noCond ). And now it no longer works (see attached screenshot) And just for completeness: There is nothing in the logs However, if I use the same two xml files on our current galaxy servers (running 15.10). It works in both cases. Each time, the right dbkey is picked up. Hans-Rudolf On 08/16/2016 01:47 PM, Hans-Rudolf Hotz wrote:
Hi Nicola
Many thanks for your comments/suggestions. I've just tested the rnaStar wrapper, and it works as well.
I will try to dig deeper
Hans-Rudolf
On 08/16/2016 01:38 PM, Nicola Soranzo wrote:
I've tested bowtie_wrapper on Galaxy release_16.04: <option type="from_data_table" name="bowtie_indexes" column="1" offset="0"> works fine for me.
Cheers, Nicola
On 16/08/16 11:42, Nicola Soranzo wrote:
Forget about my previous comment, I should have read your email better, sorry!
Your code similar to bowtie_wrapper is also used by https://github.com/galaxyproject/tools-iuc/blob/master/tools/rgrnastar/rg_rn...
and I think it should work, not sure if it's a regression in Galaxy.
Cheers, Nicola
On 16/08/16 11:29, Nicola Soranzo wrote:
Hi Hans, see my comment below.
On 16/08/16 09:51, Hans-Rudolf Hotz wrote:
Dear All
I am trying to move our Galaxy installation to new hardware and at the same time to a new version (the original is "15.10",minor version "2" via mercurial; the new is "16.04" via github):
So far, I am making good progress, but I am struggling with a tool which was written by someone else who has left more than 2 years ago. The problem is: it doesn't set the dbkey correctly.
I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
The tool xml is as follows:
<inputs> <param name="sample_file" type="data" format="qsamples,qsamplespaired" label="Sample File" help="set of sequence files created by the 'select sequence files' tool"> </param> <param name="index" type="select" label="Reference Genome" help="all reads will be mapped to this reference"> <options from_data_table="fmi__quasr_genomes"/> </param>
... </inputs> <outputs> <data name="output01" type="data" format="qproject" label="qProject of ${sample_file.name}"> <actions> <action type="metadata" name="dbkey"> <option type="from_param" name="index"/>
It seems you are missing a param_attribute="dbkey" here, it should be: <option type="from_param" name="index" param_attribute="dbkey"/>
Cheers, Nicola
</action> </actions> </data> </outputs>
The user can select a genome (based on the Bioconductor BSgenome packages) from the tab delimited fmi__quasr_genomes table, which looks like:
hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) BSgenome.Hsapiens.UCSC.hg19 hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) BSgenome.Hsapiens.UCSC.hg18 ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) BSgenome.Celegans.UCSC.ce10 ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) BSgenome.Celegans.UCSC.ce6 ...
The tool works: - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) - once the job is submitted, it uses the right Bioconductor package (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc)
However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
Has anything changed with the 'action' tag since release 15.10? which makes this simple xml no longer working?
I have to admit, it does look different than the example on the wiki (https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t...
). I tried the code from the wiki which is based on the bowtie_wrapper (https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...)
as well:
<actions> <action type="metadata" name="dbkey"> <option type="from_data_table" name="fmi__quasr_genomes" column="1" offset="0"> <filter type="param_value" column="0" value="#" compare="startswith" keep="False"/> <filter type="param_value" ref="index" column="0"/> </option> </action> </actions>
but it results in the same kind of error: <Cheetah.Template.Template object at 0x7ff82c0e43d0>
Thank you very much for any help/comments
Regards, Hans-Rudolf
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
{kind=link}

Yup - thanks for the bug report. I have used your rna example to build a minimal-ish example to fit into Galaxy's test tools framework here https://github.com/galaxyproject/galaxy/pull/2795. I also tested in 16.01 and it worked - so this clearly broke in 16.04. I'll see if I can track it down and we can move the discussion to the github PR. Thanks a bunch for the detailed report Hans! -John On Tue, Aug 16, 2016 at 9:02 AM, Hans-Rudolf Hotz <hrh@fmi.ch> wrote:
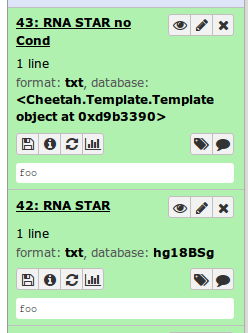
Hi Nicola and others on the list
It looks like it has something to do with the 'conditional'
I took the rnaStar xml file, and reduced it to the bare bones, just to reproduce the issue (see attachment: rg_rnaStar.simple ). This works. It picks up the right dbkey.
I made the xml file even simpler and removed the 'conditional' (see attachment: rg_rnaStar.simple_noCond ). And now it no longer works (see attached screenshot)
And just for completeness: There is nothing in the logs
However, if I use the same two xml files on our current galaxy servers (running 15.10). It works in both cases. Each time, the right dbkey is picked up.
Hans-Rudolf
On 08/16/2016 01:47 PM, Hans-Rudolf Hotz wrote:
Hi Nicola
Many thanks for your comments/suggestions. I've just tested the rnaStar wrapper, and it works as well.
I will try to dig deeper
Hans-Rudolf
On 08/16/2016 01:38 PM, Nicola Soranzo wrote:
I've tested bowtie_wrapper on Galaxy release_16.04: <option type="from_data_table" name="bowtie_indexes" column="1" offset="0"> works fine for me.
Cheers, Nicola
On 16/08/16 11:42, Nicola Soranzo wrote:
Forget about my previous comment, I should have read your email better, sorry!
Your code similar to bowtie_wrapper is also used by
https://github.com/galaxyproject/tools-iuc/blob/master/tools/rgrnastar/rg_rn...
and I think it should work, not sure if it's a regression in Galaxy.
Cheers, Nicola
On 16/08/16 11:29, Nicola Soranzo wrote:
Hi Hans, see my comment below.
On 16/08/16 09:51, Hans-Rudolf Hotz wrote:
Dear All
I am trying to move our Galaxy installation to new hardware and at the same time to a new version (the original is "15.10",minor version "2" via mercurial; the new is "16.04" via github):
So far, I am making good progress, but I am struggling with a tool which was written by someone else who has left more than 2 years ago. The problem is: it doesn't set the dbkey correctly.
I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
The tool xml is as follows:
<inputs> <param name="sample_file" type="data" format="qsamples,qsamplespaired" label="Sample File" help="set of sequence files created by the 'select sequence files' tool"> </param> <param name="index" type="select" label="Reference Genome" help="all reads will be mapped to this reference"> <options from_data_table="fmi__quasr_genomes"/> </param>
... </inputs> <outputs> <data name="output01" type="data" format="qproject" label="qProject of ${sample_file.name}"> <actions> <action type="metadata" name="dbkey"> <option type="from_param" name="index"/>
It seems you are missing a param_attribute="dbkey" here, it should be: <option type="from_param" name="index" param_attribute="dbkey"/>
Cheers, Nicola
</action> </actions> </data> </outputs>
The user can select a genome (based on the Bioconductor BSgenome packages) from the tab delimited fmi__quasr_genomes table, which looks like:
hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) BSgenome.Hsapiens.UCSC.hg19 hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) BSgenome.Hsapiens.UCSC.hg18 ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) BSgenome.Celegans.UCSC.ce10 ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) BSgenome.Celegans.UCSC.ce6 ...
The tool works: - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) - once the job is submitted, it uses the right Bioconductor package (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc)
However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: <Cheetah.Template.Template object at 0x7f1e40210f50>
Has anything changed with the 'action' tag since release 15.10? which makes this simple xml no longer working?
I have to admit, it does look different than the example on the wiki
(https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t...
). I tried the code from the wiki which is based on the bowtie_wrapper
(https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...)
as well:
<actions> <action type="metadata" name="dbkey"> <option type="from_data_table" name="fmi__quasr_genomes" column="1" offset="0"> <filter type="param_value" column="0" value="#" compare="startswith" keep="False"/> <filter type="param_value" ref="index" column="0"/> </option> </action> </actions>
but it results in the same kind of error: <Cheetah.Template.Template object at 0x7ff82c0e43d0>
Thank you very much for any help/comments
Regards, Hans-Rudolf
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
Hi John and Nicola Thank you very much for working on this and presenting a solution so fast! Hans-Rudolf PS: https://github.com/galaxyproject/galaxy/pull/2800 On 08/16/2016 04:40 PM, John Chilton wrote:
Yup - thanks for the bug report. I have used your rna example to build a minimal-ish example to fit into Galaxy's test tools framework here https://github.com/galaxyproject/galaxy/pull/2795. I also tested in 16.01 and it worked - so this clearly broke in 16.04. I'll see if I can track it down and we can move the discussion to the github PR. Thanks a bunch for the detailed report Hans!
-John
On Tue, Aug 16, 2016 at 9:02 AM, Hans-Rudolf Hotz <hrh@fmi.ch> wrote:
Hi Nicola and others on the list
It looks like it has something to do with the 'conditional'
I took the rnaStar xml file, and reduced it to the bare bones, just to reproduce the issue (see attachment: rg_rnaStar.simple ). This works. It picks up the right dbkey.
I made the xml file even simpler and removed the 'conditional' (see attachment: rg_rnaStar.simple_noCond ). And now it no longer works (see attached screenshot)
And just for completeness: There is nothing in the logs
However, if I use the same two xml files on our current galaxy servers (running 15.10). It works in both cases. Each time, the right dbkey is picked up.
Hans-Rudolf
On 08/16/2016 01:47 PM, Hans-Rudolf Hotz wrote:
Hi Nicola
Many thanks for your comments/suggestions. I've just tested the rnaStar wrapper, and it works as well.
I will try to dig deeper
Hans-Rudolf
On 08/16/2016 01:38 PM, Nicola Soranzo wrote:
I've tested bowtie_wrapper on Galaxy release_16.04: <option type="from_data_table" name="bowtie_indexes" column="1" offset="0"> works fine for me.
Cheers, Nicola
On 16/08/16 11:42, Nicola Soranzo wrote:
Forget about my previous comment, I should have read your email better, sorry!
Your code similar to bowtie_wrapper is also used by
https://github.com/galaxyproject/tools-iuc/blob/master/tools/rgrnastar/rg_rn...
and I think it should work, not sure if it's a regression in Galaxy.
Cheers, Nicola
On 16/08/16 11:29, Nicola Soranzo wrote:
Hi Hans, see my comment below.
On 16/08/16 09:51, Hans-Rudolf Hotz wrote: > > Dear All > > > I am trying to move our Galaxy installation to new hardware and at > the same time to a new version (the original is "15.10",minor > version "2" via mercurial; the new is "16.04" via github): > > So far, I am making good progress, but I am struggling with a tool > which was written by someone else who has left more than 2 years > ago. The problem is: it doesn't set the dbkey correctly. > > I get: > <Cheetah.Template.Template object at 0x7f1e40210f50> > > > > The tool xml is as follows: > > <inputs> > <param name="sample_file" type="data" > format="qsamples,qsamplespaired" label="Sample File" help="set of > sequence files created by the 'select sequence files' tool"> > </param> > <param name="index" type="select" label="Reference Genome" > help="all reads will be mapped to this reference"> > <options from_data_table="fmi__quasr_genomes"/> > </param> > > ... > </inputs> > <outputs> > <data name="output01" type="data" format="qproject" > label="qProject of ${sample_file.name}"> > <actions> > <action type="metadata" name="dbkey"> > <option type="from_param" name="index"/>
It seems you are missing a param_attribute="dbkey" here, it should be: <option type="from_param" name="index" param_attribute="dbkey"/>
Cheers, Nicola
> </action> > </actions> > </data> > </outputs> > > > > The user can select a genome (based on the Bioconductor BSgenome > packages) from the tab delimited fmi__quasr_genomes table, which > looks like: > > hg19BSg hg19BSg BSgenome Hsapiens (UCSC hg19) > BSgenome.Hsapiens.UCSC.hg19 > hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18) > BSgenome.Hsapiens.UCSC.hg18 > ce10BSg ce10BSg BSgenome Celegans (UCSC ce10) > BSgenome.Celegans.UCSC.ce10 > ce6BSg ce6BSg BSgenome Celegans (UCSC ce6) > BSgenome.Celegans.UCSC.ce6 > ... > > > The tool works: > - it displays the available genomes (BSgenome Hsapiens (UCSC hg19), > hg18BSg hg18BSg BSgenome Hsapiens (UCSC hg18), etc) > - once the job is submitted, it uses the right Bioconductor package > (BSgenome.Hsapiens.UCSC.hg19, BSgenome.Hsapiens.UCSC.hg18, etc) > > However, instead of the dbkey (hg19BSg, hg18BSg, etc) I get: > <Cheetah.Template.Template object at 0x7f1e40210f50> > > > > Has anything changed with the 'action' tag since release 15.10? > which makes this simple xml no longer working? > > I have to admit, it does look different than the example on the wiki > > (https://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Cactions.3E_t... > > ). I tried the code from the wiki which is based on the > bowtie_wrapper > > (https://github.com/galaxyproject/tools-devteam/blob/master/tools/bowtie_wrap...) > > as well: > > > <actions> > <action type="metadata" name="dbkey"> > <option type="from_data_table" name="fmi__quasr_genomes" > column="1" offset="0"> > <filter type="param_value" column="0" value="#" > compare="startswith" keep="False"/> > <filter type="param_value" ref="index" column="0"/> > </option> > </action> > </actions> > > > but it results in the same kind of error: > <Cheetah.Template.Template object at 0x7ff82c0e43d0> > > > Thank you very much for any help/comments > > Regards, Hans-Rudolf > > > > > > >
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
participants (3)
-
 Hans-Rudolf Hotz
Hans-Rudolf Hotz -
 John Chilton
John Chilton -
 Nicola Soranzo
Nicola Soranzo