why is my lastz process showing it is not finished?

I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning. Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing? David Hoover Helix Systems Staff http://helix.nih.gov

Hi all, I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off? Best, Nico --------------------------------------------------------------- Nicolas Delhomme High Throughput Functional Genomics Center European Molecular Biology Laboratory Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany --------------------------------------------------------------- On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev

Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico, If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting: set_metadata_externally = True In universe_wsgi.ini. --nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Thanks, Nico --------------------------------------------------------------- Nicolas Delhomme High Throughput Functional Genomics Center European Molecular Biology Laboratory Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany --------------------------------------------------------------- On 22 Nov 2010, at 16:21, Nate Coraor wrote:
Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico,
If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting:
set_metadata_externally = True
In universe_wsgi.ini.
--nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
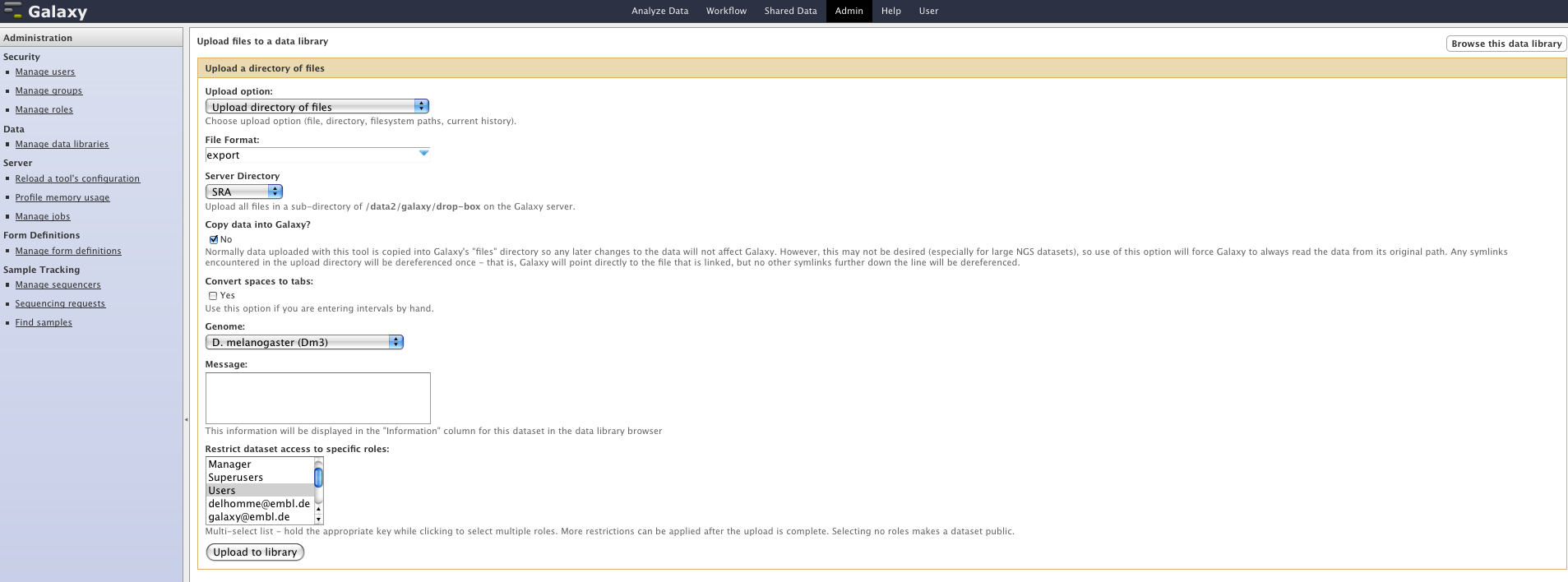
Hi Nate, I followed your instructions, creating a datatype and providing it. However, loading the files still takes ages (>1h); I do it as an admin, going through the following menu: Admin>Manage data libraries>[my library(GAIIx)]>Add datasets>Upload directory of files. See the attached screenshot for the parameters I set (Picture2.png). Any idea what could be the reason for it? I've attached the content of the mysql job table for that job (job.sql.txt). Here is my datatype definition in the corresponding files (datatypes_conf.xml, registry.py and tabular.py). Look for the Export Class or the export keyword. Btw the export format is an Illumina default standard, which you might want to have in Galaxy as a default :-) I haven't set the metadata information yet, but could have a look at it if you find this datatype useful and would consider adding it as a default. Thanks for your help, Nico --------------------------------------------------------------- Nicolas Delhomme High Throughput Functional Genomics Center European Molecular Biology Laboratory Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany --------------------------------------------------------------- On 22 Nov 2010, at 16:21, Nate Coraor wrote:
Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico,
If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting:
set_metadata_externally = True
In universe_wsgi.ini.
--nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
{kind=link}
Nicolas Delhomme wrote:
Hi Nate,
I followed your instructions, creating a datatype and providing it. However, loading the files still takes ages (>1h); I do it as an admin, going through the following menu: Admin>Manage data libraries>[my library(GAIIx)]>Add datasets>Upload directory of files. See the attached screenshot for the parameters I set (Picture2.png).
Any idea what could be the reason for it? I've attached the content of the mysql job table for that job (job.sql.txt).
Here is my datatype definition in the corresponding files (datatypes_conf.xml, registry.py and tabular.py). Look for the Export Class or the export keyword.
Btw the export format is an Illumina default standard, which you might want to have in Galaxy as a default :-) I haven't set the metadata information yet, but could have a look at it if you find this datatype useful and would consider adding it as a default.
Hi Nico, Quick question, have you configured: set_metadata_externally = True In universe_wsgi.ini? My immediate thought is that since Export is subclassed from Tabular, it is attempting to determine the type (int/float/list/string) of the content of every column on every row in the file. This is expensive on large files, but unfortunately whenever we've tried to set a maximum number of lines to check in the past, that maximum has been too low. If your file format is guaranteed to be able to yield the correct types within X number of lines (or you don't care that incorrect determination of column types would break things like the Filter tool), you should define set_meta() in your custom type and have it call Tabular.set_meta() with max_data_lines = X. To see an example of max_data_lines in use, have a look at datatypes.interval:Wiggle.set_meta(). --nate
<?xml version="1.0"?> <datatypes> <registration converters_path="lib/galaxy/datatypes/converters" display_path="display_applications"> <datatype extension="ab1" type="galaxy.datatypes.binary:Ab1" mimetype="application/octet-stream" display_in_upload="true"/> <datatype extension="axt" type="galaxy.datatypes.sequence:Axt" display_in_upload="true"/> <datatype extension="bam" type="galaxy.datatypes.binary:Bam" mimetype="application/octet-stream" display_in_upload="true"> <converter file="bam_to_bai.xml" target_datatype="bai"/> <!-- <converter file="bam_to_array_tree_converter.xml" target_datatype="array_tree"/> --> <display file="ucsc/bam.xml" /> </datatype> <datatype extension="bed" type="galaxy.datatypes.interval:Bed" display_in_upload="true"> <converter file="bed_to_gff_converter.xml" target_datatype="gff"/> <converter file="interval_to_coverage.xml" target_datatype="coverage"/> <converter file="bed_to_interval_index_converter.xml" target_datatype="interval_index"/> <!-- <converter file="bed_to_array_tree_converter.xml" target_datatype="array_tree"/> --> <converter file="bed_to_genetrack_converter.xml" target_datatype="genetrack"/> <!-- <display file="ucsc/interval_as_bed.xml" /> --> <display file="genetrack.xml" /> </datatype> <datatype extension="bedstrict" type="galaxy.datatypes.interval:BedStrict" /> <!-- <datatype extension="binseq.zip" type="galaxy.datatypes.binary:Binseq" mimetype="application/zip" display_in_upload="true"/> --> <datatype extension="len" type="galaxy.datatypes.chrominfo:ChromInfo" display_in_upload="true"> <!-- no converters yet --> </datatype> <datatype extension="coverage" type="galaxy.datatypes.coverage:LastzCoverage" display_in_upload="true"> <indexer file="coverage.xml" /> </datatype> <datatype extension="customtrack" type="galaxy.datatypes.interval:CustomTrack"/> <datatype extension="csfasta" type="galaxy.datatypes.sequence:csFasta" display_in_upload="true"/> <datatype extension="data" type="galaxy.datatypes.data:Data" mimetype="application/octet-stream"/> <datatype extension="export" type="galaxy.datatypes.tabular:Export" display_in_upload="true"/> <datatype extension="fasta" type="galaxy.datatypes.sequence:Fasta" display_in_upload="true"> <converter file="fasta_to_tabular_converter.xml" target_datatype="tabular"/> </datatype> <datatype extension="fastq" type="galaxy.datatypes.sequence:Fastq" display_in_upload="true"/> <datatype extension="fastqsanger" type="galaxy.datatypes.sequence:FastqSanger" display_in_upload="true"/> <datatype extension="fastqsolexa" type="galaxy.datatypes.sequence:FastqSolexa" display_in_upload="true"/> <datatype extension="fastqcssanger" type="galaxy.datatypes.sequence:FastqCSSanger" display_in_upload="true"/> <datatype extension="fastqillumina" type="galaxy.datatypes.sequence:FastqIllumina" display_in_upload="true"/> <datatype extension="genetrack" type="galaxy.datatypes.tracks:GeneTrack"> <!-- <display file="genetrack.xml" /> --> </datatype> <datatype extension="gff" type="galaxy.datatypes.interval:Gff" display_in_upload="true"> <converter file="gff_to_bed_converter.xml" target_datatype="bed"/> </datatype> <datatype extension="gff3" type="galaxy.datatypes.interval:Gff3" display_in_upload="true"/> <datatype extension="gif" type="galaxy.datatypes.images:Image" mimetype="image/gif"/> <datatype extension="gmaj.zip" type="galaxy.datatypes.images:Gmaj" mimetype="application/zip"/> <datatype extension="html" type="galaxy.datatypes.images:Html" mimetype="text/html"/> <datatype extension="interval" type="galaxy.datatypes.interval:Interval" display_in_upload="true"> <converter file="interval_to_bed_converter.xml" target_datatype="bed"/> <converter file="interval_to_bedstrict_converter.xml" target_datatype="bedstrict"/> <indexer file="interval_awk.xml" /> <!-- <display file="ucsc/interval_as_bed.xml" inherit="True" /> --> <display file="genetrack.xml" inherit="True"/> </datatype> <datatype extension="jpg" type="galaxy.datatypes.images:Image" mimetype="image/jpeg"/> <datatype extension="laj" type="galaxy.datatypes.images:Laj"/> <datatype extension="lav" type="galaxy.datatypes.sequence:Lav" display_in_upload="true"/> <datatype extension="maf" type="galaxy.datatypes.sequence:Maf" display_in_upload="true"> <converter file="maf_to_fasta_converter.xml" target_datatype="fasta"/> <converter file="maf_to_interval_converter.xml" target_datatype="interval"/> </datatype> <datatype extension="pdf" type="galaxy.datatypes.images:Image" mimetype="application/pdf"/> <datatype extension="png" type="galaxy.datatypes.images:Image" mimetype="image/png"/> <datatype extension="qual" type="galaxy.datatypes.qualityscore:QualityScore" /> <datatype extension="qualsolexa" type="galaxy.datatypes.qualityscore:QualityScoreSolexa" display_in_upload="true"/> <datatype extension="qualillumina" type="galaxy.datatypes.qualityscore:QualityScoreIllumina" display_in_upload="true"/> <datatype extension="qualsolid" type="galaxy.datatypes.qualityscore:QualityScoreSOLiD" display_in_upload="true"/> <datatype extension="qual454" type="galaxy.datatypes.qualityscore:QualityScore454" display_in_upload="true"/> <datatype extension="sam" type="galaxy.datatypes.tabular:Sam" display_in_upload="true"/> <datatype extension="scf" type="galaxy.datatypes.binary:Scf" mimetype="application/octet-stream" display_in_upload="true"/> <datatype extension="sff" type="galaxy.datatypes.binary:Sff" mimetype="application/octet-stream" display_in_upload="true"/> <datatype extension="taxonomy" type="galaxy.datatypes.tabular:Taxonomy" display_in_upload="true"/> <datatype extension="tabular" type="galaxy.datatypes.tabular:Tabular" display_in_upload="true"/> <datatype extension="txt" type="galaxy.datatypes.data:Text" display_in_upload="true"/> <datatype extension="blastxml" type="galaxy.datatypes.xml:BlastXml" display_in_upload="true"/> <!-- <datatype extension="txtseq.zip" type="galaxy.datatypes.data:Txtseq" mimetype="application/zip" display_in_upload="true"/> --> <datatype extension="wig" type="galaxy.datatypes.interval:Wiggle" display_in_upload="true"> <converter file="wiggle_to_array_tree_converter.xml" target_datatype="array_tree"/> <converter file="wiggle_to_simple_converter.xml" target_datatype="interval"/> </datatype> <datatype extension="array_tree" type="galaxy.datatypes.data:Data" /> <datatype extension="interval_index" type="galaxy.datatypes.data:Data" /> <!-- Start EMBOSS tools --> <datatype extension="acedb" type="galaxy.datatypes.data:Text"/> <datatype extension="asn1" type="galaxy.datatypes.data:Text"/> <datatype extension="btwisted" type="galaxy.datatypes.data:Text"/> <datatype extension="cai" type="galaxy.datatypes.data:Text"/> <datatype extension="charge" type="galaxy.datatypes.data:Text"/> <datatype extension="checktrans" type="galaxy.datatypes.data:Text"/> <datatype extension="chips" type="galaxy.datatypes.data:Text"/> <datatype extension="clustal" type="galaxy.datatypes.data:Text"/> <datatype extension="codata" type="galaxy.datatypes.data:Text"/> <datatype extension="codcmp" type="galaxy.datatypes.data:Text"/> <datatype extension="coderet" type="galaxy.datatypes.data:Text"/> <datatype extension="compseq" type="galaxy.datatypes.data:Text"/> <datatype extension="cpgplot" type="galaxy.datatypes.data:Text"/> <datatype extension="cpgreport" type="galaxy.datatypes.data:Text"/> <datatype extension="cusp" type="galaxy.datatypes.data:Text"/> <datatype extension="cut" type="galaxy.datatypes.data:Text"/> <datatype extension="dan" type="galaxy.datatypes.data:Text"/> <datatype extension="dbmotif" type="galaxy.datatypes.data:Text"/> <datatype extension="diffseq" type="galaxy.datatypes.data:Text"/> <datatype extension="digest" type="galaxy.datatypes.data:Text"/> <datatype extension="dreg" type="galaxy.datatypes.data:Text"/> <datatype extension="einverted" type="galaxy.datatypes.data:Text"/> <datatype extension="embl" type="galaxy.datatypes.data:Text"/> <datatype extension="epestfind" type="galaxy.datatypes.data:Text"/> <datatype extension="equicktandem" type="galaxy.datatypes.data:Text"/> <datatype extension="est2genome" type="galaxy.datatypes.data:Text"/> <datatype extension="etandem" type="galaxy.datatypes.data:Text"/> <datatype extension="excel" type="galaxy.datatypes.data:Text"/> <datatype extension="feattable" type="galaxy.datatypes.data:Text"/> <datatype extension="fitch" type="galaxy.datatypes.data:Text"/> <datatype extension="freak" type="galaxy.datatypes.data:Text"/> <datatype extension="fuzznuc" type="galaxy.datatypes.data:Text"/> <datatype extension="fuzzpro" type="galaxy.datatypes.data:Text"/> <datatype extension="fuzztran" type="galaxy.datatypes.data:Text"/> <datatype extension="garnier" type="galaxy.datatypes.data:Text"/> <datatype extension="gcg" type="galaxy.datatypes.data:Text"/> <datatype extension="geecee" type="galaxy.datatypes.data:Text"/> <datatype extension="genbank" type="galaxy.datatypes.data:Text"/> <datatype extension="helixturnhelix" type="galaxy.datatypes.data:Text"/> <datatype extension="hennig86" type="galaxy.datatypes.data:Text"/> <datatype extension="hmoment" type="galaxy.datatypes.data:Text"/> <datatype extension="ig" type="galaxy.datatypes.data:Text"/> <datatype extension="isochore" type="galaxy.datatypes.data:Text"/> <datatype extension="jackknifer" type="galaxy.datatypes.data:Text"/> <datatype extension="jackknifernon" type="galaxy.datatypes.data:Text"/> <datatype extension="markx10" type="galaxy.datatypes.data:Text"/> <datatype extension="markx1" type="galaxy.datatypes.data:Text"/> <datatype extension="markx0" type="galaxy.datatypes.data:Text"/> <datatype extension="markx3" type="galaxy.datatypes.data:Text"/> <datatype extension="markx2" type="galaxy.datatypes.data:Text"/> <datatype extension="match" type="galaxy.datatypes.data:Text"/> <datatype extension="mega" type="galaxy.datatypes.data:Text"/> <datatype extension="meganon" type="galaxy.datatypes.data:Text"/> <datatype extension="motif" type="galaxy.datatypes.data:Text"/> <datatype extension="msf" type="galaxy.datatypes.data:Text"/> <datatype extension="nametable" type="galaxy.datatypes.data:Text"/> <datatype extension="ncbi" type="galaxy.datatypes.data:Text"/> <datatype extension="needle" type="galaxy.datatypes.data:Text"/> <datatype extension="newcpgreport" type="galaxy.datatypes.data:Text"/> <datatype extension="newcpgseek" type="galaxy.datatypes.data:Text"/> <datatype extension="nexus" type="galaxy.datatypes.data:Text"/> <datatype extension="nexusnon" type="galaxy.datatypes.data:Text"/> <datatype extension="noreturn" type="galaxy.datatypes.data:Text"/> <datatype extension="pair" type="galaxy.datatypes.data:Text"/> <datatype extension="palindrome" type="galaxy.datatypes.data:Text"/> <datatype extension="pepcoil" type="galaxy.datatypes.data:Text"/> <datatype extension="pepinfo" type="galaxy.datatypes.data:Text"/> <datatype extension="pepstats" type="galaxy.datatypes.data:Text"/> <datatype extension="phylip" type="galaxy.datatypes.data:Text"/> <datatype extension="phylipnon" type="galaxy.datatypes.data:Text"/> <datatype extension="pir" type="galaxy.datatypes.data:Text"/> <datatype extension="polydot" type="galaxy.datatypes.data:Text"/> <datatype extension="preg" type="galaxy.datatypes.data:Text"/> <datatype extension="prettyseq" type="galaxy.datatypes.data:Text"/> <datatype extension="primersearch" type="galaxy.datatypes.data:Text"/> <datatype extension="regions" type="galaxy.datatypes.data:Text"/> <datatype extension="score" type="galaxy.datatypes.data:Text"/> <datatype extension="selex" type="galaxy.datatypes.data:Text"/> <datatype extension="seqtable" type="galaxy.datatypes.data:Text"/> <datatype extension="showfeat" type="galaxy.datatypes.data:Text"/> <datatype extension="showorf" type="galaxy.datatypes.data:Text"/> <datatype extension="simple" type="galaxy.datatypes.data:Text"/> <datatype extension="sixpack" type="galaxy.datatypes.data:Text"/> <datatype extension="srs" type="galaxy.datatypes.data:Text"/> <datatype extension="srspair" type="galaxy.datatypes.data:Text"/> <datatype extension="staden" type="galaxy.datatypes.data:Text"/> <datatype extension="strider" type="galaxy.datatypes.data:Text"/> <datatype extension="supermatcher" type="galaxy.datatypes.data:Text"/> <datatype extension="swiss" type="galaxy.datatypes.data:Text"/> <datatype extension="syco" type="galaxy.datatypes.data:Text"/> <datatype extension="table" type="galaxy.datatypes.data:Text"/> <datatype extension="textsearch" type="galaxy.datatypes.data:Text"/> <datatype extension="vectorstrip" type="galaxy.datatypes.data:Text"/> <datatype extension="wobble" type="galaxy.datatypes.data:Text"/> <datatype extension="wordcount" type="galaxy.datatypes.data:Text"/> <datatype extension="tagseq" type="galaxy.datatypes.data:Text"/> <!-- End EMBOSS tools --> <!-- Start RGenetics Datatypes --> <datatype extension="affybatch" type="galaxy.datatypes.genetics:Affybatch" display_in_upload="true"/> <!-- eigenstrat pedigree input file --> <datatype extension="eigenstratgeno" type="galaxy.datatypes.genetics:Eigenstratgeno"/> <!-- eigenstrat pca output file for adjusted eigenQTL eg --> <datatype extension="eigenstratpca" type="galaxy.datatypes.genetics:Eigenstratpca"/> <datatype extension="eset" type="galaxy.datatypes.genetics:Eset" display_in_upload="true" /> <!-- fbat/pbat format pedigree (header row of marker names) --> <datatype extension="fped" type="galaxy.datatypes.genetics:Fped" display_in_upload="true"/> <!-- phenotype file - fbat format --> <datatype extension="fphe" type="galaxy.datatypes.genetics:Fphe" display_in_upload="true" mimetype="text/html"/> <!-- genome graphs ucsc file - first col is always marker then numeric values to plot --> <datatype extension="gg" type="galaxy.datatypes.genetics:GenomeGraphs"/> <!-- part of linkage format pedigree --> <datatype extension="malist" type="galaxy.datatypes.genetics:MAlist" display_in_upload="true"/> <!-- linkage format pedigree (separate .map file) --> <datatype extension="lped" type="galaxy.datatypes.genetics:Lped" display_in_upload="true"> <converter file="lped_to_fped_converter.xml" target_datatype="fped"/> <converter file="lped_to_pbed_converter.xml" target_datatype="pbed"/> </datatype> <!-- plink compressed file - has bed extension unfortunately --> <datatype extension="pbed" type="galaxy.datatypes.genetics:Pbed" display_in_upload="true"> <converter file="pbed_to_lped_converter.xml" target_datatype="lped"/> </datatype> <datatype extension="pheno" type="galaxy.datatypes.genetics:Pheno"/> <!-- phenotype file - plink format --> <datatype extension="pphe" type="galaxy.datatypes.genetics:Pphe" display_in_upload="true" mimetype="text/html"/> <datatype extension="rexpbase" type="galaxy.datatypes.genetics:RexpBase"/> <datatype extension="rgenetics" type="galaxy.datatypes.genetics:Rgenetics"/> <datatype extension="snptest" type="galaxy.datatypes.genetics:Snptest" display_in_upload="true"/> <datatype extension="snpmatrix" type="galaxy.datatypes.genetics:SNPMatrix" display_in_upload="true"/> <datatype extension="xls" type="galaxy.datatypes.tabular:Tabular"/> <!-- End RGenetics Datatypes --> </registration> <sniffers> <!-- The order in which Galaxy attempts to determine data types is important because some formats are much more loosely defined than others. The following list should be the most rigidly defined format first, followed by next-most rigidly defined, and so on. --> <sniffer type="galaxy.datatypes.binary:Bam"/> <sniffer type="galaxy.datatypes.binary:Sff"/> <sniffer type="galaxy.datatypes.xml:BlastXml"/> <sniffer type="galaxy.datatypes.sequence:Maf"/> <sniffer type="galaxy.datatypes.sequence:Lav"/> <sniffer type="galaxy.datatypes.sequence:csFasta"/> <sniffer type="galaxy.datatypes.qualityscore:QualityScoreSOLiD"/> <sniffer type="galaxy.datatypes.qualityscore:QualityScore454"/> <sniffer type="galaxy.datatypes.sequence:Fasta"/> <sniffer type="galaxy.datatypes.sequence:Fastq"/> <sniffer type="galaxy.datatypes.interval:Wiggle"/> <sniffer type="galaxy.datatypes.images:Html"/> <sniffer type="galaxy.datatypes.sequence:Axt"/> <sniffer type="galaxy.datatypes.interval:Bed"/> <sniffer type="galaxy.datatypes.interval:CustomTrack"/> <sniffer type="galaxy.datatypes.interval:Gff"/> <sniffer type="galaxy.datatypes.interval:Gff3"/> <sniffer type="galaxy.datatypes.interval:Interval"/> <sniffer type="galaxy.datatypes.tabular:Sam"/> <sniffer type="galaxy.datatypes.tabular:Export"/> </sniffers> </datatypes>
""" Provides mapping between extensions and datatypes, mime-types, etc. """ import os, tempfile import logging import data, tabular, interval, images, sequence, qualityscore, genetics, xml, coverage, tracks, chrominfo, binary, assembly, ngsindex import galaxy.util from galaxy.util.odict import odict from display_applications.application import DisplayApplication
class ConfigurationError( Exception ): pass
class Registry( object ): def __init__( self, root_dir=None, config=None ): self.log = logging.getLogger(__name__) self.log.addHandler( logging.NullHandler() ) self.datatypes_by_extension = {} self.mimetypes_by_extension = {} self.datatype_converters = odict() self.datatype_indexers = odict() self.converters = [] self.converter_deps = {} self.available_tracks = [] self.set_external_metadata_tool = None self.indexers = [] self.sniff_order = [] self.upload_file_formats = [] self.display_applications = odict() #map a display application id to a display application inherit_display_application_by_class = [] if root_dir and config: # Parse datatypes_conf.xml tree = galaxy.util.parse_xml( config ) root = tree.getroot() # Load datatypes and converters from config self.log.debug( 'Loading datatypes from %s' % config ) registration = root.find( 'registration' ) self.datatype_converters_path = os.path.join( root_dir, registration.get( 'converters_path', 'lib/galaxy/datatypes/converters' ) ) self.datatype_indexers_path = os.path.join( root_dir, registration.get( 'indexers_path', 'lib/galaxy/datatypes/indexers' ) ) self.display_applications_path = os.path.join( root_dir, registration.get( 'display_path', 'display_applications' ) ) if not os.path.isdir( self.datatype_converters_path ): raise ConfigurationError( "Directory does not exist: %s" % self.datatype_converters_path ) if not os.path.isdir( self.datatype_indexers_path ): raise ConfigurationError( "Directory does not exist: %s" % self.datatype_indexers_path ) for elem in registration.findall( 'datatype' ): try: extension = elem.get( 'extension', None ) dtype = elem.get( 'type', None ) mimetype = elem.get( 'mimetype', None ) display_in_upload = elem.get( 'display_in_upload', False ) if extension and dtype: fields = dtype.split( ':' ) datatype_module = fields[0] datatype_class = fields[1] fields = datatype_module.split( '.' ) module = __import__( fields.pop(0) ) for mod in fields: module = getattr( module, mod ) self.datatypes_by_extension[extension] = getattr( module, datatype_class )() if mimetype is None: # Use default mime type as per datatype spec mimetype = self.datatypes_by_extension[extension].get_mime() self.mimetypes_by_extension[extension] = mimetype if hasattr( getattr( module, datatype_class ), "get_track_type" ): self.available_tracks.append( extension ) if display_in_upload: self.upload_file_formats.append( extension ) #max file size cut off for setting optional metadata self.datatypes_by_extension[extension].max_optional_metadata_filesize = elem.get( 'max_optional_metadata_filesize', None ) for converter in elem.findall( 'converter' ): # Build the list of datatype converters which will later be loaded # into the calling app's toolbox. converter_config = converter.get( 'file', None ) target_datatype = converter.get( 'target_datatype', None ) depends_on = converter.get( 'depends_on', None ) if depends_on and target_datatype: if extension not in self.converter_deps: self.converter_deps[extension] = {} self.converter_deps[extension][target_datatype] = depends_on.split(',') if converter_config and target_datatype: self.converters.append( ( converter_config, extension, target_datatype ) ) for indexer in elem.findall( 'indexer' ): # Build the list of datatype indexers for track building indexer_config = indexer.get( 'file', None ) if indexer_config: self.indexers.append( (indexer_config, extension) ) for composite_file in elem.findall( 'composite_file' ): # add composite files name = composite_file.get( 'name', None ) if name is None: self.log.warning( "You must provide a name for your composite_file (%s)." % composite_file ) optional = composite_file.get( 'optional', False ) mimetype = composite_file.get( 'mimetype', None ) self.datatypes_by_extension[extension].add_composite_file( name, optional=optional, mimetype=mimetype ) for display_app in elem.findall( 'display' ): display_file = os.path.join( self.display_applications_path, display_app.get( 'file', None ) ) try: inherit = galaxy.util.string_as_bool( display_app.get( 'inherit', 'False' ) ) display_app = DisplayApplication.from_file( display_file, self ) if display_app: if display_app.id in self.display_applications: #if we already loaded this display application, we'll use the first one again display_app = self.display_applications[ display_app.id ] self.log.debug( "Loaded display application '%s' for datatype '%s', inherit=%s" % ( display_app.id, extension, inherit ) ) self.display_applications[ display_app.id ] = display_app #Display app by id self.datatypes_by_extension[ extension ].add_display_application( display_app ) if inherit and ( self.datatypes_by_extension[extension], display_app ) not in inherit_display_application_by_class: #subclass inheritance will need to wait until all datatypes have been loaded inherit_display_application_by_class.append( ( self.datatypes_by_extension[extension], display_app ) ) except: self.log.exception( "error reading display application from path: %s" % display_file ) except Exception, e: self.log.warning( 'Error loading datatype "%s", problem: %s' % ( extension, str( e ) ) ) # Handle display_application subclass inheritance here: for ext, d_type1 in self.datatypes_by_extension.iteritems(): for d_type2, display_app in inherit_display_application_by_class: current_app = d_type1.get_display_application( display_app.id, None ) if current_app is None and isinstance( d_type1, type( d_type2 ) ): d_type1.add_display_application( display_app ) # Load datatype sniffers from the config sniff_order = [] sniffers = root.find( 'sniffers' ) for elem in sniffers.findall( 'sniffer' ): dtype = elem.get( 'type', None ) if dtype: sniff_order.append( dtype ) for dtype in sniff_order: try: fields = dtype.split( ":" ) datatype_module = fields[0] datatype_class = fields[1] fields = datatype_module.split( "." ) module = __import__( fields.pop(0) ) for mod in fields: module = getattr( module, mod ) aclass = getattr( module, datatype_class )() included = False for atype in self.sniff_order: if not issubclass( atype.__class__, aclass.__class__ ) and isinstance( atype, aclass.__class__ ): included = True break if not included: self.sniff_order.append( aclass ) self.log.debug( 'Loaded sniffer for datatype: %s' % dtype ) except Exception, exc: self.log.warning( 'Error appending datatype %s to sniff_order, problem: %s' % ( dtype, str( exc ) ) ) #default values if len(self.datatypes_by_extension) < 1: self.datatypes_by_extension = { 'ab1' : binary.Ab1(), 'axt' : sequence.Axt(), 'bam' : binary.Bam(), 'bed' : interval.Bed(), 'blastxml' : xml.BlastXml(), 'coverage' : coverage.LastzCoverage(), 'customtrack' : interval.CustomTrack(), 'csfasta' : sequence.csFasta(), 'export' : tabular.Export(), 'fasta' : sequence.Fasta(), 'fastq' : sequence.Fastq(), 'fastqsanger' : sequence.FastqSanger(), 'gtf' : interval.Gtf(), 'gff' : interval.Gff(), 'gff3' : interval.Gff3(), 'genetrack' : tracks.GeneTrack(), 'interval' : interval.Interval(), 'laj' : images.Laj(), 'lav' : sequence.Lav(), 'maf' : sequence.Maf(), 'pileup' : tabular.Pileup(), 'qualsolid' : qualityscore.QualityScoreSOLiD(), 'qualsolexa' : qualityscore.QualityScoreSolexa(), 'qual454' : qualityscore.QualityScore454(), 'sam' : tabular.Sam(), 'scf' : binary.Scf(), 'sff' : binary.Sff(), 'tabular' : tabular.Tabular(), 'taxonomy' : tabular.Taxonomy(), 'txt' : data.Text(), 'wig' : interval.Wiggle() } self.mimetypes_by_extension = { 'ab1' : 'application/octet-stream', 'axt' : 'text/plain', 'bam' : 'application/octet-stream', 'bed' : 'text/plain', 'blastxml' : 'text/plain', 'customtrack' : 'text/plain', 'csfasta' : 'text/plain', 'export' : 'text/plain', 'fasta' : 'text/plain', 'fastq' : 'text/plain', 'fastqsanger' : 'text/plain', 'gtf' : 'text/plain', 'gff' : 'text/plain', 'gff3' : 'text/plain', 'interval' : 'text/plain', 'laj' : 'text/plain', 'lav' : 'text/plain', 'maf' : 'text/plain', 'pileup' : 'text/plain', 'qualsolid' : 'text/plain', 'qualsolexa' : 'text/plain', 'qual454' : 'text/plain', 'sam' : 'text/plain', 'scf' : 'application/octet-stream', 'sff' : 'application/octet-stream', 'tabular' : 'text/plain', 'taxonomy' : 'text/plain', 'txt' : 'text/plain', 'wig' : 'text/plain' } # super supertype fix for input steps in workflows. if 'data' not in self.datatypes_by_extension: self.datatypes_by_extension['data'] = data.Data() self.mimetypes_by_extension['data'] = 'application/octet-stream' # Default values - the order in which we attempt to determine data types is critical # because some formats are much more flexibly defined than others. if len(self.sniff_order) < 1: self.sniff_order = [ binary.Bam(), binary.Sff(), xml.BlastXml(), sequence.Maf(), sequence.Lav(), sequence.csFasta(), qualityscore.QualityScoreSOLiD(), qualityscore.QualityScore454(), sequence.Fasta(), sequence.Fastq(), interval.Wiggle(), images.Html(), sequence.Axt(), interval.Bed(), interval.CustomTrack(), interval.Gtf(), interval.Gff(), interval.Gff3(), tabular.Pileup(), interval.Interval(), tabular.Sam(), tabular.Export() ] def append_to_sniff_order(): # Just in case any supported data types are not included in the config's sniff_order section. for ext in self.datatypes_by_extension: datatype = self.datatypes_by_extension[ext] included = False for atype in self.sniff_order: if isinstance(atype, datatype.__class__): included = True break if not included: self.sniff_order.append(datatype) append_to_sniff_order()
def get_available_tracks(self): return self.available_tracks
def get_mimetype_by_extension(self, ext, default = 'application/octet-stream' ): """Returns a mimetype based on an extension""" try: mimetype = self.mimetypes_by_extension[ext] except KeyError: #datatype was never declared mimetype = default self.log.warning('unknown mimetype in data factory %s' % ext) return mimetype
def get_datatype_by_extension(self, ext ): """Returns a datatype based on an extension""" try: builder = self.datatypes_by_extension[ext] except KeyError: builder = data.Text() return builder
def change_datatype(self, data, ext, set_meta = True ): data.extension = ext # call init_meta and copy metadata from itself. The datatype # being converted *to* will handle any metadata copying and # initialization. if data.has_data(): data.set_size() data.init_meta( copy_from=data ) if set_meta: #metadata is being set internally data.set_meta( overwrite = False ) data.set_peek() return data
def old_change_datatype(self, data, ext): """Creates and returns a new datatype based on an existing data and an extension""" newdata = factory(ext)(id=data.id) for key, value in data.__dict__.items(): setattr(newdata, key, value) newdata.ext = ext return newdata
def load_datatype_converters( self, toolbox ): """Adds datatype converters from self.converters to the calling app's toolbox""" for elem in self.converters: tool_config = elem[0] source_datatype = elem[1] target_datatype = elem[2] converter_path = os.path.join( self.datatype_converters_path, tool_config ) try: converter = toolbox.load_tool( converter_path ) toolbox.tools_by_id[converter.id] = converter if source_datatype not in self.datatype_converters: self.datatype_converters[source_datatype] = odict() self.datatype_converters[source_datatype][target_datatype] = converter self.log.debug( "Loaded converter: %s", converter.id ) except: self.log.exception( "error reading converter from path: %s" % converter_path )
def load_external_metadata_tool( self, toolbox ): """Adds a tool which is used to set external metadata""" #we need to be able to add a job to the queue to set metadata. The queue will currently only accept jobs with an associated tool. #We'll create a special tool to be used for Auto-Detecting metadata; this is less than ideal, but effective #Properly building a tool without relying on parsing an XML file is near impossible...so we'll create a temporary file tool_xml_text = """ <tool id="__SET_METADATA__" name="Set External Metadata" version="1.0.1" tool_type="set_metadata"> <type class="SetMetadataTool" module="galaxy.tools"/> <action module="galaxy.tools.actions.metadata" class="SetMetadataToolAction"/> <command>$__SET_EXTERNAL_METADATA_COMMAND_LINE__</command> <inputs> <param format="data" name="input1" type="data" label="File to set metadata on."/> <param name="__ORIGINAL_DATASET_STATE__" type="hidden" value=""/> <param name="__SET_EXTERNAL_METADATA_COMMAND_LINE__" type="hidden" value=""/> </inputs> </tool> """ tmp_name = tempfile.NamedTemporaryFile() tmp_name.write( tool_xml_text ) tmp_name.flush() set_meta_tool = toolbox.load_tool( tmp_name.name ) toolbox.tools_by_id[ set_meta_tool.id ] = set_meta_tool self.set_external_metadata_tool = set_meta_tool self.log.debug( "Loaded external metadata tool: %s", self.set_external_metadata_tool.id )
def load_datatype_indexers( self, toolbox ): """Adds indexers from self.indexers to the toolbox from app""" for elem in self.indexers: tool_config = elem[0] datatype = elem[1] indexer = toolbox.load_tool( os.path.join( self.datatype_indexers_path, tool_config ) ) toolbox.tools_by_id[indexer.id] = indexer self.datatype_indexers[datatype] = indexer self.log.debug( "Loaded indexer: %s", indexer.id )
def get_converters_by_datatype(self, ext): """Returns available converters by source type""" converters = odict() source_datatype = type(self.get_datatype_by_extension(ext)) for ext2, dict in self.datatype_converters.items(): converter_datatype = type(self.get_datatype_by_extension(ext2)) if issubclass(source_datatype, converter_datatype): converters.update(dict) #Ensure ext-level converters are present if ext in self.datatype_converters.keys(): converters.update(self.datatype_converters[ext]) return converters
def get_indexers_by_datatype( self, ext ): """Returns indexers based on datatype""" class_chain = list() source_datatype = type(self.get_datatype_by_extension(ext)) for ext_spec in self.datatype_indexers.keys(): datatype = type(self.get_datatype_by_extension(ext_spec)) if issubclass( source_datatype, datatype ): class_chain.append( ext_spec ) # Prioritize based on class chain ext2type = lambda x: self.get_datatype_by_extension(x) class_chain = sorted(class_chain, lambda x,y: issubclass(ext2type(x),ext2type(y)) and -1 or 1) return [self.datatype_indexers[x] for x in class_chain]
def get_converter_by_target_type(self, source_ext, target_ext): """Returns a converter based on source and target datatypes""" converters = self.get_converters_by_datatype(source_ext) if target_ext in converters.keys(): return converters[target_ext] return None
def find_conversion_destination_for_dataset_by_extensions( self, dataset, accepted_formats, converter_safe = True ): """Returns ( target_ext, existing converted dataset )""" for convert_ext in self.get_converters_by_datatype( dataset.ext ): if isinstance( self.get_datatype_by_extension( convert_ext ), accepted_formats ): dataset = dataset.get_converted_files_by_type( convert_ext ) if dataset: ret_data = dataset elif not converter_safe: continue else: ret_data = None return ( convert_ext, ret_data ) return ( None, None )
def get_composite_extensions( self ): return [ ext for ( ext, d_type ) in self.datatypes_by_extension.iteritems() if d_type.composite_type is not None ]
def get_upload_metadata_params( self, context, group, tool ): """Returns dict of case value:inputs for metadata conditional for upload tool""" rval = {} for ext, d_type in self.datatypes_by_extension.iteritems(): inputs = [] for meta_name, meta_spec in d_type.metadata_spec.iteritems(): if meta_spec.set_in_upload: help_txt = meta_spec.desc if not help_txt or help_txt == meta_name: help_txt = "" inputs.append( '<param type="text" name="%s" label="Set metadata value for "%s"" value="%s" help="%s"/>' % ( meta_name, meta_name, meta_spec.default, help_txt ) ) rval[ ext ] = "\n".join( inputs ) if 'auto' not in rval and 'txt' in rval: #need to manually add 'auto' datatype rval[ 'auto' ] = rval[ 'txt' ] return rval
""" Tabular datatype
""" import pkg_resources pkg_resources.require( "bx-python" )
import logging import data from galaxy import util from cgi import escape from galaxy.datatypes import metadata from galaxy.datatypes.metadata import MetadataElement import galaxy_utils.sequence.vcf from sniff import *
log = logging.getLogger(__name__)
class Tabular( data.Text ): """Tab delimited data"""
"""Add metadata elements""" MetadataElement( name="comment_lines", default=0, desc="Number of comment lines", readonly=False, optional=True, no_value=0 ) MetadataElement( name="columns", default=0, desc="Number of columns", readonly=True, visible=False, no_value=0 ) MetadataElement( name="column_types", default=[], desc="Column types", param=metadata.ColumnTypesParameter, readonly=True, visible=False, no_value=[] )
def init_meta( self, dataset, copy_from=None ): data.Text.init_meta( self, dataset, copy_from=copy_from ) def set_meta( self, dataset, overwrite = True, skip = None, max_data_lines = None, **kwd ): """ Tries to determine the number of columns as well as those columns that contain numerical values in the dataset. A skip parameter is used because various tabular data types reuse this function, and their data type classes are responsible to determine how many invalid comment lines should be skipped. Using None for skip will cause skip to be zero, but the first line will be processed as a header. A max_data_lines parameter is used because various tabular data types reuse this function, and their data type classes are responsible to determine how many data lines should be processed to ensure that the non-optional metadata parameters are properly set; if used, optional metadata parameters will be set to None, unless the entire file has already been read. Using None (default) for max_data_lines will process all data lines.
Items of interest: 1. We treat 'overwrite' as always True (we always want to set tabular metadata when called). 2. If a tabular file has no data, it will have one column of type 'str'. 3. We used to check only the first 100 lines when setting metadata and this class's set_peek() method read the entire file to determine the number of lines in the file. Since metadata can now be processed on cluster nodes, we've merged the line count portion of the set_peek() processing here, and we now check the entire contents of the file. """ # Store original skip value to check with later requested_skip = skip if skip is None: skip = 0 column_type_set_order = [ 'int', 'float', 'list', 'str' ] #Order to set column types in default_column_type = column_type_set_order[-1] # Default column type is lowest in list column_type_compare_order = list( column_type_set_order ) #Order to compare column types column_type_compare_order.reverse() def type_overrules_type( column_type1, column_type2 ): if column_type1 is None or column_type1 == column_type2: return False if column_type2 is None: return True for column_type in column_type_compare_order: if column_type1 == column_type: return True if column_type2 == column_type: return False #neither column type was found in our ordered list, this cannot happen raise "Tried to compare unknown column types" def is_int( column_text ): try: int( column_text ) return True except: return False def is_float( column_text ): try: float( column_text ) return True except: if column_text.strip().lower() == 'na': return True #na is special cased to be a float return False def is_list( column_text ): return "," in column_text def is_str( column_text ): #anything, except an empty string, is True if column_text == "": return False return True is_column_type = {} #Dict to store column type string to checking function for column_type in column_type_set_order: is_column_type[column_type] = locals()[ "is_%s" % ( column_type ) ] def guess_column_type( column_text ): for column_type in column_type_set_order: if is_column_type[column_type]( column_text ): return column_type return None data_lines = 0 comment_lines = 0 column_types = [] first_line_column_types = [default_column_type] # default value is one column of type str if dataset.has_data(): #NOTE: if skip > num_check_lines, we won't detect any metadata, and will use default dataset_fh = open( dataset.file_name ) i = 0 while True: line = dataset_fh.readline() if not line: break line = line.rstrip( '\r\n' ) if i < skip or not line or line.startswith( '#' ): # We'll call blank lines comments comment_lines += 1 else: data_lines += 1 fields = line.split( '\t' ) for field_count, field in enumerate( fields ): if field_count >= len( column_types ): #found a previously unknown column, we append None column_types.append( None ) column_type = guess_column_type( field ) if type_overrules_type( column_type, column_types[field_count] ): column_types[field_count] = column_type if i == 0 and requested_skip is None: # This is our first line, people seem to like to upload files that have a header line, but do not # start with '#' (i.e. all column types would then most likely be detected as str). We will assume # that the first line is always a header (this was previous behavior - it was always skipped). When # the requested skip is None, we only use the data from the first line if we have no other data for # a column. This is far from perfect, as # 1,2,3 1.1 2.2 qwerty # 0 0 1,2,3 # will be detected as # "column_types": ["int", "int", "float", "list"] # instead of # "column_types": ["list", "float", "float", "str"] *** would seem to be the 'Truth' by manual # observation that the first line should be included as data. The old method would have detected as # "column_types": ["int", "int", "str", "list"] first_line_column_types = column_types column_types = [ None for col in first_line_column_types ] if max_data_lines is not None and data_lines >= max_data_lines: if dataset_fh.tell() != dataset.get_size(): data_lines = None #Clear optional data_lines metadata value comment_lines = None #Clear optional comment_lines metadata value; additional comment lines could appear below this point break i += 1 dataset_fh.close()
#we error on the larger number of columns #first we pad our column_types by using data from first line if len( first_line_column_types ) > len( column_types ): for column_type in first_line_column_types[len( column_types ):]: column_types.append( column_type ) #Now we fill any unknown (None) column_types with data from first line for i in range( len( column_types ) ): if column_types[i] is None: if len( first_line_column_types ) <= i or first_line_column_types[i] is None: column_types[i] = default_column_type else: column_types[i] = first_line_column_types[i] # Set the discovered metadata values for the dataset dataset.metadata.data_lines = data_lines dataset.metadata.comment_lines = comment_lines dataset.metadata.column_types = column_types dataset.metadata.columns = len( column_types ) def make_html_table( self, dataset, skipchars=[] ): """Create HTML table, used for displaying peek""" out = ['<table cellspacing="0" cellpadding="3">'] try: out.append( '<tr>' ) # Generate column header for i in range( 1, dataset.metadata.columns+1 ): out.append( '<th>%s</th>' % str( i ) ) out.append( '</tr>' ) out.append( self.make_html_peek_rows( dataset, skipchars=skipchars ) ) out.append( '</table>' ) out = "".join( out ) except Exception, exc: out = "Can't create peek %s" % str( exc ) return out def make_html_peek_rows( self, dataset, skipchars=[] ): out = [""] comments = [] if not dataset.peek: dataset.set_peek() data = dataset.peek lines = data.splitlines() for line in lines: line = line.rstrip( '\r\n' ) if not line: continue comment = False for skipchar in skipchars: if line.startswith( skipchar ): comments.append( line ) comment = True break if comment: continue elems = line.split( '\t' ) if len( elems ) != dataset.metadata.columns: # We may have an invalid comment line or invalid data comments.append( line ) comment = True continue while len( comments ) > 0: # Keep comments try: out.append( '<tr><td colspan="100%">' ) except: out.append( '<tr><td>' ) out.append( '%s</td></tr>' % escape( comments.pop(0) ) ) out.append( '<tr>' ) for elem in elems: # valid data elem = escape( elem ) out.append( '<td>%s</td>' % elem ) out.append( '</tr>' ) # Peek may consist only of comments while len( comments ) > 0: try: out.append( '<tr><td colspan="100%">' ) except: out.append( '<tr><td>' ) out.append( '%s</td></tr>' % escape( comments.pop(0) ) ) return "".join( out ) def set_peek( self, dataset, line_count=None, is_multi_byte=False ): data.Text.set_peek( self, dataset, line_count=line_count, is_multi_byte=is_multi_byte ) if dataset.metadata.comment_lines: dataset.blurb = "%s, %s comments" % ( dataset.blurb, util.commaify( str( dataset.metadata.comment_lines ) ) ) def display_peek( self, dataset ): """Returns formatted html of peek""" return self.make_html_table( dataset ) def displayable( self, dataset ): try: return dataset.has_data() \ and dataset.state == dataset.states.OK \ and dataset.metadata.columns > 0 \ and dataset.metadata.data_lines > 0 except: return False def as_gbrowse_display_file( self, dataset, **kwd ): return open( dataset.file_name ) def as_ucsc_display_file( self, dataset, **kwd ): return open( dataset.file_name )
class Taxonomy( Tabular ): def __init__(self, **kwd): """Initialize taxonomy datatype""" Tabular.__init__( self, **kwd ) self.column_names = ['Name', 'TaxId', 'Root', 'Superkingdom', 'Kingdom', 'Subkingdom', 'Superphylum', 'Phylum', 'Subphylum', 'Superclass', 'Class', 'Subclass', 'Superorder', 'Order', 'Suborder', 'Superfamily', 'Family', 'Subfamily', 'Tribe', 'Subtribe', 'Genus', 'Subgenus', 'Species', 'Subspecies' ] def make_html_table( self, dataset, skipchars=[] ): """Create HTML table, used for displaying peek""" out = ['<table cellspacing="0" cellpadding="3">'] comments = [] try: # Generate column header out.append( '<tr>' ) for i, name in enumerate( self.column_names ): out.append( '<th>%s.%s</th>' % ( str( i+1 ), name ) ) # This data type requires at least 24 columns in the data if dataset.metadata.columns - len( self.column_names ) > 0: for i in range( len( self.column_names ), dataset.metadata.columns ): out.append( '<th>%s</th>' % str( i+1 ) ) out.append( '</tr>' ) out.append( self.make_html_peek_rows( dataset, skipchars=skipchars ) ) out.append( '</table>' ) out = "".join( out ) except Exception, exc: out = "Can't create peek %s" % exc return out
class Sam( Tabular ): file_ext = 'sam' def __init__(self, **kwd): """Initialize taxonomy datatype""" Tabular.__init__( self, **kwd ) self.column_names = ['QNAME', 'FLAG', 'RNAME', 'POS', 'MAPQ', 'CIGAR', 'MRNM', 'MPOS', 'ISIZE', 'SEQ', 'QUAL', 'OPT' ] def make_html_table( self, dataset, skipchars=[] ): """Create HTML table, used for displaying peek""" out = ['<table cellspacing="0" cellpadding="3">'] try: # Generate column header out.append( '<tr>' ) for i, name in enumerate( self.column_names ): out.append( '<th>%s.%s</th>' % ( str( i+1 ), name ) ) # This data type requires at least 11 columns in the data if dataset.metadata.columns - len( self.column_names ) > 0: for i in range( len( self.column_names ), dataset.metadata.columns ): out.append( '<th>%s</th>' % str( i+1 ) ) out.append( '</tr>' ) out.append( self.make_html_peek_rows( dataset, skipchars=skipchars ) ) out.append( '</table>' ) out = "".join( out ) except Exception, exc: out = "Can't create peek %s" % exc return out def sniff( self, filename ): """ Determines whether the file is in SAM format
A file in SAM format consists of lines of tab-separated data. The following header line may be the first line: @QNAME FLAG RNAME POS MAPQ CIGAR MRNM MPOS ISIZE SEQ QUAL or @QNAME FLAG RNAME POS MAPQ CIGAR MRNM MPOS ISIZE SEQ QUAL OPT Data in the OPT column is optional and can consist of tab-separated data
For complete details see http://samtools.sourceforge.net/SAM1.pdf
Rules for sniffing as True: There must be 11 or more columns of data on each line Columns 2 (FLAG), 4(POS), 5 (MAPQ), 8 (MPOS), and 9 (ISIZE) must be numbers (9 can be negative) We will only check that up to the first 5 alignments are correctly formatted.
>>> fname = get_test_fname( 'sequence.maf' ) >>> Sam().sniff( fname ) False >>> fname = get_test_fname( '1.sam' ) >>> Sam().sniff( fname ) True """ try: fh = open( filename ) count = 0 while True: line = fh.readline() line = line.strip() if not line: break #EOF if line: if line[0] != '@': linePieces = line.split('\t') if len(linePieces) < 11: return False try: check = int(linePieces[1]) check = int(linePieces[3]) check = int(linePieces[4]) check = int(linePieces[7]) check = int(linePieces[8]) except ValueError: return False count += 1 if count == 5: return True fh.close() if count < 5 and count > 0: return True except: pass return False
class Export( Tabular ): file_ext = 'export' def __init__(self, **kwd): """Initialize export datatype""" Tabular.__init__( self, **kwd ) self.column_names = ['MACHINE', 'RUN', 'LANE', 'TILE', 'X', 'Y', 'MULTIPLEX', 'PAIRID', 'READ', 'QUALITY', 'CHROMOSOME', 'CONTIG', 'POSITION','STRAND','ALN_QUAL','CHASTITY' ] def make_html_table( self, dataset, skipchars=[] ): """Create HTML table, used for displaying peek""" out = ['<table cellspacing="0" cellpadding="3">'] try: # Generate column header out.append( '<tr>' ) for i, name in enumerate( self.column_names ): out.append( '<th>%s.%s</th>' % ( str( i+1 ), name ) ) # This data type requires at least 16 columns in the data if dataset.metadata.columns - len( self.column_names ) > 0: for i in range( len( self.column_names ), dataset.metadata.columns ): out.append( '<th>%s</th>' % str( i+1 ) ) out.append( '</tr>' ) out.append( self.make_html_peek_rows( dataset, skipchars=skipchars ) ) out.append( '</table>' ) out = "".join( out ) except Exception, exc: out = "Can't create peek %s" % exc return out def sniff( self, filename ): """ Determines whether the file is in Export format
A file in Export format consists of lines of tab-separated data. It does not have any header
Rules for sniffing as True: There must be 16 columns of data on each line Columns 2 to 8 must be numbers Column 16 should be either Y or N We will only check that up to the first 5 alignments are correctly formatted.
""" try: fh = open( filename ) count = 0 while True: line = fh.readline() line = line.strip() if not line: break #EOF if line: if line[0] != '@': linePieces = line.split('\t') if len(linePieces) != 16: return False try: check = int(linePieces[1]) check = int(linePieces[2]) check = int(linePieces[3]) check = int(linePieces[4]) check = int(linePieces[5]) check = int(linePieces[6]) check = int(linePieces[7]) assert linePieces[15] in [ 'Y', 'N' ] except ValueError: return False count += 1 if count == 5: return True fh.close() if count < 5 and count > 0: return True except: pass return False
class Pileup( Tabular ): """Tab delimited data in pileup (6- or 10-column) format""" file_ext = "pileup"
"""Add metadata elements""" MetadataElement( name="chromCol", default=1, desc="Chrom column", param=metadata.ColumnParameter ) MetadataElement( name="startCol", default=2, desc="Start column", param=metadata.ColumnParameter ) MetadataElement( name="baseCol", default=3, desc="Reference base column", param=metadata.ColumnParameter )
def init_meta( self, dataset, copy_from=None ): Tabular.init_meta( self, dataset, copy_from=copy_from )
def set_peek( self, dataset, line_count=None, is_multi_byte=False ): """Set the peek and blurb text""" if not dataset.dataset.purged: dataset.peek = data.get_file_peek( dataset.file_name, is_multi_byte=is_multi_byte ) if line_count is None: # See if line_count is stored in the metadata if dataset.metadata.data_lines: dataset.blurb = "%s genomic coordinates" % util.commaify( str( dataset.metadata.data_lines ) ) else: # Number of lines is not known ( this should not happen ), and auto-detect is # needed to set metadata dataset.blurb = "? genomic coordinates" else: dataset.blurb = "%s genomic coordinates" % util.commaify( str( line_count ) ) else: dataset.peek = 'file does not exist' dataset.blurb = 'file purged from disk'
def make_html_table( self, dataset, skipchars=[] ): """Create HTML table, used for displaying peek""" out = ['<table cellspacing="0" cellpadding="3">'] comments = [] try: # Generate column header out.append('<tr>') for i in range( 1, dataset.metadata.columns+1 ): if i == dataset.metadata.chromCol: out.append( '<th>%s.Chrom</th>' % i ) elif i == dataset.metadata.startCol: out.append( '<th>%s.Start</th>' % i ) elif i == dataset.metadata.baseCol: out.append( '<th>%s.Base</th>' % i ) else: out.append( '<th>%s</th>' % i ) out.append('</tr>') out.append( self.make_html_peek_rows( dataset, skipchars=skipchars ) ) out.append( '</table>' ) out = "".join( out ) except Exception, exc: out = "Can't create peek %s" % str( exc ) return out
def repair_methods( self, dataset ): """Return options for removing errors along with a description""" return [ ("lines", "Remove erroneous lines") ]
def sniff( self, filename ): """ Checks for 'pileup-ness'
There are two main types of pileup: 6-column and 10-column. For both, the first three and last two columns are the same. We only check the first three to allow for some personalization of the format.
>>> fname = get_test_fname( 'interval.interval' ) >>> Pileup().sniff( fname ) False >>> fname = get_test_fname( '6col.pileup' ) >>> Pileup().sniff( fname ) True >>> fname = get_test_fname( '10col.pileup' ) >>> Pileup().sniff( fname ) True """ headers = get_headers( filename, '\t' ) try: for hdr in headers: if hdr and not hdr[0].startswith( '#' ): if len( hdr ) < 3: return False try: # chrom start in column 1 (with 0-based columns) # and reference base is in column 2 check = int( hdr[1] ) assert hdr[2] in [ 'A', 'C', 'G', 'T', 'N', 'a', 'c', 'g', 't', 'n' ] except: return False return True except: return False
class Eland( Tabular ): file_ext = 'eland'
def sniff( self, filename ): return False
class ElandMulti( Tabular ): file_ext = 'elandmulti'
def sniff( self, filename ): return False
class Vcf( Tabular ): """ Variant Call Format for describing SNPs and other simple genome variations. """
file_ext = 'vcf' column_names = [ 'Chrom', 'Pos', 'ID', 'Ref', 'Alt', 'Qual', 'Filter', 'Info', 'Format', 'data' ]
MetadataElement( name="columns", default=10, desc="Number of columns", readonly=True, visible=False ) MetadataElement( name="column_types", default=['str','int','str','str','str','int','str','list','str','str'], param=metadata.ColumnTypesParameter, desc="Column types", readonly=True, visible=False ) MetadataElement( name="viz_filter_cols", default=[5], param=metadata.ColumnParameter, multiple=True )
def sniff( self, filename ): try: # If reader can read and parse file, it's VCF. for line in list( galaxy_utils.sequence.vcf.Reader( open( filename ) ) ): pass return True except: return False
def make_html_table( self, dataset, skipchars=[] ): """Create HTML table, used for displaying peek""" out = ['<table cellspacing="0" cellpadding="3">'] try: # Generate column header out.append( '<tr>' ) for i, name in enumerate( self.column_names ): out.append( '<th>%s.%s</th>' % ( str( i+1 ), name ) ) out.append( self.make_html_peek_rows( dataset, skipchars=skipchars ) ) out.append( '</table>' ) out = "".join( out ) except Exception, exc: out = "Can't create peek %s" % exc return out
def get_track_type( self ): return "FeatureTrack", {"data": "interval_index", "index": "summary_tree"}
id create_time update_time history_id tool_id tool_version state info command_line param_filename runner_name stdout stderr traceback session_id job_runner_name job_runner_external_id library_folder_id user_id imported 2674 2010-11-25 10:19:08 2010-11-25 10:19:10 (null) upload1 1.1.2 running (null) python /home/galaxy/galaxy_dev/tools/data_source/upload.py /home/galaxy/galaxy_dev /home/galaxy/galaxy_dev/datatypes_conf.xml /tmp/tmpeNuQgu 3501:/home/galaxy/galaxy_dev/database/job_working_directory/2674/dataset_3501_files:/data2/galaxy/drop-box/GAIIx/s_4.export 3502:/home/galaxy/galaxy_dev/database/job_working_directory/2674/dataset_3502_files:/data2/galaxy/drop-box/GAIIx/s_6.export 3503:/home/galaxy/galaxy_dev/database/job_working_directory/2674/dataset_3503_files:/data2/galaxy/drop-box/GAIIx/s_7.export 3504:/home/galaxy/galaxy_dev/database/job_working_directory/2674/dataset_3504_files:/data2/galaxy/drop-box/GAIIx/s_8.export (null) (null) (null) (null) (null) 143 local:/// 17003 48 1 false
Thanks for your help,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 22 Nov 2010, at 16:21, Nate Coraor wrote:
Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico,
If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting:
set_metadata_externally = True
In universe_wsgi.ini.
--nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Hi Nate, Thanks, that did the trick. Are you interested to have that file format in Galaxy? Just let me know and I'll send you a proper diff of the different files. Cheers, Nico --------------------------------------------------------------- Nicolas Delhomme High Throughput Functional Genomics Center European Molecular Biology Laboratory Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany --------------------------------------------------------------- On 22 Nov 2010, at 16:21, Nate Coraor wrote:
Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico,
If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting:
set_metadata_externally = True
In universe_wsgi.ini.
--nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev

Nicolas: Do you also happen to have export2fastq converter? Yes, it is simple, but if you have it already we would put into Format conversion section as many people inquired about it. Thanks! anton On Dec 2, 2010, at 6:57 AM, Nicolas Delhomme wrote:
Hi Nate,
Thanks, that did the trick.
Are you interested to have that file format in Galaxy? Just let me know and I'll send you a proper diff of the different files.
Cheers,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 22 Nov 2010, at 16:21, Nate Coraor wrote:
Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico,
If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting:
set_metadata_externally = True
In universe_wsgi.ini.
--nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Anton Nekrutenko http://nekrut.bx.psu.edu http://usegalaxy.org
Hi Anton, Yes, I do. I'll create a bundle with everything and send it to you. Cheers, Nico --------------------------------------------------------------- Nicolas Delhomme High Throughput Functional Genomics Center European Molecular Biology Laboratory Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany --------------------------------------------------------------- On 2 Dec 2010, at 14:45, Anton Nekrutenko wrote:
Nicolas:
Do you also happen to have export2fastq converter? Yes, it is simple, but if you have it already we would put into Format conversion section as many people inquired about it.
Thanks!
anton
On Dec 2, 2010, at 6:57 AM, Nicolas Delhomme wrote:
Hi Nate,
Thanks, that did the trick.
Are you interested to have that file format in Galaxy? Just let me know and I'll send you a proper diff of the different files.
Cheers,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 22 Nov 2010, at 16:21, Nate Coraor wrote:
Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico,
If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting:
set_metadata_externally = True
In universe_wsgi.ini.
--nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Anton Nekrutenko http://nekrut.bx.psu.edu http://usegalaxy.org
Nico: Once you have the bundle put it into http://usegalaxy.org/community and we'll take from there. Thanks! a. On Dec 2, 2010, at 8:53 AM, Nicolas Delhomme wrote:
Hi Anton,
Yes, I do. I'll create a bundle with everything and send it to you.
Cheers,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 2 Dec 2010, at 14:45, Anton Nekrutenko wrote:
Nicolas:
Do you also happen to have export2fastq converter? Yes, it is simple, but if you have it already we would put into Format conversion section as many people inquired about it.
Thanks!
anton
On Dec 2, 2010, at 6:57 AM, Nicolas Delhomme wrote:
Hi Nate,
Thanks, that did the trick.
Are you interested to have that file format in Galaxy? Just let me know and I'll send you a proper diff of the different files.
Cheers,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 22 Nov 2010, at 16:21, Nate Coraor wrote:
Nicolas Delhomme wrote:
Hi all,
I do see the see the same, whenever loading export (one of Illumina sequencer output) files (as an admin uploading a directory of files) or when running bowtie/bwa/samtools. I'm wondering if this would have something to do with the Sniffers (i.e. that you're validating the generated files)? Actually as I'm running a local Galaxy instance and am sure that the format are correct, being directly at the source, I would not need such validations. Is there a way to turn that off?
Nico,
If you set the file type on the upload form, the datatype will not be sniffed. In addition, setting the rest of the metadata can be sped up by setting:
set_metadata_externally = True
In universe_wsgi.ini.
--nate
Best,
Nico
--------------------------------------------------------------- Nicolas Delhomme
High Throughput Functional Genomics Center
European Molecular Biology Laboratory
Tel: +49 6221 387 8310 Email: nicolas.delhomme@embl.de Meyerhofstrasse 1 - Postfach 10.2209 69102 Heidelberg, Germany ---------------------------------------------------------------
On 10 Nov 2010, at 21:08, David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Anton Nekrutenko http://nekrut.bx.psu.edu http://usegalaxy.org
Anton Nekrutenko http://nekrut.bx.psu.edu http://usegalaxy.org
David Hoover wrote:
I've set up a local instance of Galaxy, and have submitted a lastz job on some fake data. It ran for about 3 minutes and then finished, as shown in the logfiles and by system tools like top and ps. However, the web page shows the spinning wheel and yellow box for 10 minutes until the box turned green and wheel stopped spinning.
Why is there such a delay between when the job completes and when the web page shows it is finished? Is there a configuration element I'm missing?
It's setting metadata. This process can probably be sped up by setting: set_metadata_externally = True In universe_wsgi.ini. We are probably going to make this the default shortly.
David Hoover Helix Systems Staff http://helix.nih.gov _______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
participants (4)
-
 Anton Nekrutenko
Anton Nekrutenko -
 David Hoover
David Hoover -
 Nate Coraor
Nate Coraor -
 Nicolas Delhomme
Nicolas Delhomme