
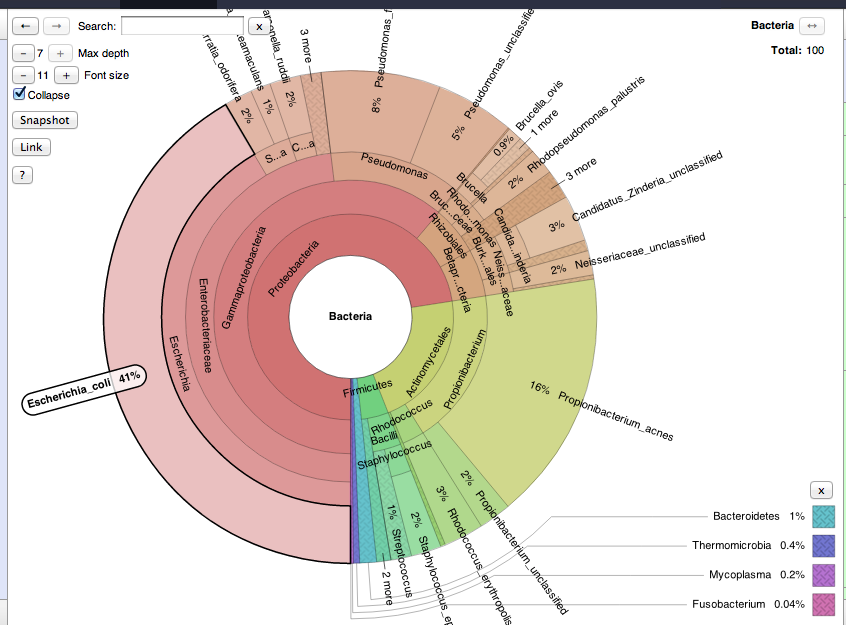
Well.. it turned out to be pretty easy thanks to the efforts of Dannon and Brian Ondov I've added the metaphlan2krona.py script to the metaphlan toolshed repo. diff -r e88fa24fa837 metaphlan.xml --- a/metaphlan.xml Wed Jun 06 10:41:23 2012 -0400 +++ b/metaphlan.xml Wed Aug 22 21:36:21 2012 -0400 @@ -14,7 +14,7 @@ </command> <inputs> - <param format="fasta" name="input" type="data" label="Input metagenome (multi-fasta of metagenomic reads, loaded with the Get Data module, see below for an example)"></param> + <param format="fasta,fastq,fastqsanger" name="input" type="data" label="Input metagenome (multi-fasta of metagenomic reads, loaded with the Get Data module, see below for an example)"></param> <param name="PresetsForBowtie2" type="select" format="text"> <label>Sensitivity options for read-marker similarity (as described by BowTie2)</label> <option value="very-sensitive-local">Very Sensitive Local</option> diff -r e88fa24fa837 metaphlan2krona.py --- /dev/null Thu Jan 01 00:00:00 1970 +0000 +++ b/metaphlan2krona.py Wed Aug 22 21:36:21 2012 -0400 @@ -0,0 +1,49 @@ +#!/usr/bin/env python + +# ============================================================================== +# Conversion script: from MetaPhlAn output to Krona text input file +# Author: Daniel Brami (daniel.brami@gmail.com<mailto:daniel.brami@gmail.com>) +# ============================================================================== + +import sys +import optparse +import re + +def main(): + #Parse Command Line + parser = optparse.OptionParser() + parser.add_option( '-p', '--profile', dest='profile', default='', action='store', help='The input file is the MetaPhlAn standard result file' ) + parser.add_option( '-k', '--krona', dest='krona', default='krona.out', action='store', help='the Krons output file name' ) + ( options, spillover ) = parser.parse_args() + + if not options.profile or not options.krona: + parser.print_help() + sys.exit() + + re_candidates = re.compile(r"s__|unclassified\t") + re_replace = re.compile(r"\w__") + re_bar = re.compile(r"\|") + + metaPhLan = list() + with open(options.profile,'r') as f: + metaPhLan = f.readlines() + f.close() + + krona_tmp = options.krona + metaPhLan_FH = open(krona_tmp, 'w') + + for aline in (metaPhLan): + if(re.search(re_candidates, aline)): + x=re.sub(re_replace, '\t', aline) + x=re.sub(re_bar, '', x) + + x_cells = x.split('\t') + lineage = '\t'.join(x_cells[0:(len(x_cells) -1)]) + abundance = float(x_cells[-1].rstrip('\n')) + + metaPhLan_FH.write('%s\n'%(str(abundance) + '\t' + lineage)) + + metaPhLan_FH.close() + +if __name__ == '__main__': + main() diff -r e88fa24fa837 metaphlan_to_krona.xml --- /dev/null Thu Jan 01 00:00:00 1970 +0000 +++ b/metaphlan_to_krona.xml Wed Aug 22 21:36:21 2012 -0400 @@ -0,0 +1,17 @@ +<tool id="meta_to_krona" name="MetaPhlAn to Krona" version="1.0.0"> + <description>Converter</description> + <command interpreter="python"> +metaphlan2krona.py -p $input -k $output + </command> + <inputs> + <param name="input" type="data" format="tabular" label="Input MetaPhlAn File"/> + </inputs> + <outputs> + <data format="tabular" name="output" label="${tool.name} on ${on_string}" /> + </outputs> + <tests> + </tests> + <help> + MetaPhlAn to Krona Converter + </help> +</tool> To get it to work with krona i extended the krona repo like this diff -r 42c899125802 krona/krona.xml --- a/krona/krona.xml Mon Mar 19 17:39:59 2012 -0400 +++ b/krona/krona.xml Wed Aug 22 21:39:17 2012 -0400 @@ -7,15 +7,18 @@ ktImportBLAST -e ${type.factor} ${type.random} + #else if $type.program == 'krona': + ktImportText #else ktImportTaxonomy ${type.summary} #end if - -o $output + -o $output ${type.include} + #unless $type.program == 'krona' + -d $depth + #end unless - -d $depth - ## uncomment for isolated intranets (see README) ## ##-u /static/krona @@ -36,6 +39,7 @@ <option value="galaxy">Galaxy taxonomic representation</option> <option value="blast">Tabular BLAST results</option> <option value="taxonomy">Taxonomy ID list</option> + <option value="krona">Krona format</option> </param> <when value="galaxy"> <param name="factor" type="hidden" value=""/> @@ -51,6 +55,10 @@ <param name="summary" type="boolean" truevalue="-m 1" falsevalue="" label="Summarized" help="The first column is counts rather than query IDs."/> <param name="include" type="boolean" truevalue="-i" falsevalue="" label="Include reads with no hits"/> </when> + <when value="krona"> + <param name="factor" type="hidden" value=""/> + <param name="include" type="hidden" value=""/> + </when> </conditional> <param name="depth" label="Depth" type="integer" help="The maximum depth to show in the chart (0 for unlimited)." value="0"/> <conditional name="datasets"> now i see this kind of graph [cid:D121407D-64B6-4D03-AE23-9DC53B52B5DC@home.langhorst.com] one more item.. had to add sanitize_all_html = False to universe_wsgi.ini brad -- Brad Langhorst langhorst@neb.com<mailto:langhorst@neb.com> 978-380-7564
{kind=link}
participants (1)
-
 Langhorst, Brad
Langhorst, Brad