
Dear Galaxy dev team, I try to use a tool allowing to sort a bam file with the "samtools sort -n" command. This tool works on BAM test files but when I run it on a real BAM file (of 3.5 G), I got an error named "error" (see attachment) ... this does not help me. In the handler.log, the job is finished normally and I don't find the error line named "error". When I run the galaxy command directly on the server, it works. Is there a log file to follow more precisely the errors? Indeed, I don't find where the "error" comes from and so I can't resolve my problem. Here my log files : more galaxy_20175.e [bam_sort_core] merging from 34 files... more galaxy_20175.ec 0 more galaxy_20175.o [bam_index_core] the alignment is not sorted (HISEQ:123:HBFN8ADXX:1:1101:1341:3979): 24-th chr > 13-th chr [bam_index_build2] fail to index the BAM file. These "errors" are not the problem, because I have the same on the BAM test files and they give no problem (I have a green box at the end of the run, only for tests data of course). Moreover, I got the following lines in my wrapper to be sure that they are only warnings : <stdio> <regex match="fail to index" source="both" level="warning" description="Warning: fail to index the bam file" /> <regex match="the alignment is not sorted" source="both" level="warning" description="Warning: the alignment is not sorted" /> </stdio> more metadata_results_HistoryDatasetAssociation_35900_PbjjDh [true, "Metadata has been set successfully"] more handler0.log galaxy.jobs DEBUG 2015-03-03 10:06:25,824 (20175) Working directory for job is: /galaxy_data/job_working_directory/020/20175 galaxy.jobs.handler DEBUG 2015-03-03 10:06:25,850 (20175) Dispatching to drmaa runner galaxy.jobs DEBUG 2015-03-03 10:06:25,943 (20175) Persisting job destination (destination id: sge) galaxy.jobs.handler INFO 2015-03-03 10:06:25,994 (20175) Job dispatched galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:26,584 (20175) submitting file /galaxy_data/job_working_directory/020/20175/galaxy_20175.sh galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:26,584 (20175) command is: samtools sort -n /galaxy_data/files/028/dataset_28339.dat /galaxy_data/files/028/dataset_28631.dat ; mv /galaxy_data/files/028/dataset_28631.dat.bam /galaxy_data/files/028/dataset_28631.dat; return_code=$?; cd /data12/galaxy/galaxy-env/galaxy; /data12/galaxy/galaxy-env/galaxy/set_metadata.sh /galaxy_data/files /galaxy_data/job_working_directory/020/20175 . /data12/galaxy/galaxy-env/galaxy/universe_wsgi.ini /galaxy_data/tmp/tmp28hp1G /galaxy_data/job_working_directory/020/20175/galaxy.json /galaxy_data/job_working_directory/020/20175/metadata_in_HistoryDatasetAssociation_35900_vmNjk7,/galaxy_data/job_working_directory/020/20175/metadata_kwds_HistoryDatasetAssociation_35900_IHm6c9,/galaxy_data/job_working_directory/020/20175/metadata_out_HistoryDatasetAssociation_35900_CboGKT,/galaxy_data/job_working_directory/020/20175/metadata_results_HistoryDatasetAssociation_35900_PbjjDh,,/galaxy_data/job_working_directory/020/20175/metadata_override_HistoryDatasetAssociation_35900_XD1a0c; sh -c "exit $return_code" galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:26,585 (20175) native specification is: -q galaxy galaxy.jobs.runners.drmaa INFO 2015-03-03 10:06:26,818 (20175) queued as 160573 galaxy.jobs DEBUG 2015-03-03 10:06:26,874 (20175) Persisting job destination (destination id: sge) galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:27,907 (20175/160573) state change: job is queued and active galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:35,538 (20175/160573) state change: job is running galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:12:10,200 (20173/160571) state change: job finished normally galaxy.datatypes.metadata DEBUG 2015-03-03 10:12:10,451 loading metadata from file for: HistoryDatasetAssociation 35898 galaxy.jobs DEBUG 2015-03-03 10:12:10,547 job 20173 ended galaxy.datatypes.metadata DEBUG 2015-03-03 10:12:10,551 Cleaning up external metadata files galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:46:16,161 (20175/160573) state change: job finished normally galaxy.jobs DEBUG 2015-03-03 10:46:16,371 setting dataset state to ERROR galaxy.jobs DEBUG 2015-03-03 10:46:16,464 job 20175 ended I don't know why galaxy give the "setting dataset state to ERROR" line. In attachment, you can see "21 : Bam sorted" on the test file and the "20 : Sort BAM on data 17" on the real file. You can see that it's not the "[bam_index_core] the alignment is not sorted (HISEQ:123:HBFN8ADXX:1:1101:1341:3979): 24-th chr > 13-th chr [bam_index_build2] fail to index the BAM file." line which generate the error. Thank you in advance for your help. Have a nice day. Best regards, Amandine Velt Plateforme Biopuces et Séquençage +33388653529
{kind=link}

So it looks like you are sorting by read name, and then Galaxy is going to attempt to index the bam file using the command-line below: % samtools sort -n tophat_out1h.bam -o - > test.bam % samtools index test.bam test.bam.bai [bam_index_core] the alignment is not sorted (test_mRNA_5_197_46): 166
54 in 1-th chr [bam_index_build2] fail to index the BAM file.
I think Galaxy may want all tool outputs to be sorted by coordinate and not read name. Is it possible that sorting by read name and sorting by coordinate produce identical ouptut in your smaller test data but not in your larger file? Not sure if there is a work around for where a difference sorting is desired - maybe you have to implement a new datatype? We are wading into bioinformatics stuffs I don't really know much about - hopefully someone more knowledgeable will comment. -John On Wed, Mar 4, 2015 at 5:38 AM, Amandine VELT <velt@igbmc.fr> wrote:
Dear Galaxy dev team,
I try to use a tool allowing to sort a bam file with the "samtools sort -n" command. This tool works on BAM test files but when I run it on a real BAM file (of 3.5 G), I got an error named "error" (see attachment) ... this does not help me.
In the handler.log, the job is finished normally and I don't find the error line named "error". When I run the galaxy command directly on the server, it works. Is there a log file to follow more precisely the errors? Indeed, I don't find where the "error" comes from and so I can't resolve my problem.
Here my log files :
more galaxy_20175.e
[bam_sort_core] merging from 34 files...
more galaxy_20175.ec
0
more galaxy_20175.o
[bam_index_core] the alignment is not sorted (HISEQ:123:HBFN8ADXX:1:1101:1341:3979): 24-th chr > 13-th chr [bam_index_build2] fail to index the BAM file.
These "errors" are not the problem, because I have the same on the BAM test files and they give no problem (I have a green box at the end of the run, only for tests data of course). Moreover, I got the following lines in my wrapper to be sure that they are only warnings :
<stdio> <regex match="fail to index" source="both" level="warning" description="Warning: fail to index the bam file" /> <regex match="the alignment is not sorted" source="both" level="warning" description="Warning: the alignment is not sorted" /> </stdio>
more metadata_results_HistoryDatasetAssociation_35900_PbjjDh
[true, "Metadata has been set successfully"]
more handler0.log
galaxy.jobs DEBUG 2015-03-03 10:06:25,824 (20175) Working directory for job is: /galaxy_data/job_working_directory/020/20175 galaxy.jobs.handler DEBUG 2015-03-03 10:06:25,850 (20175) Dispatching to drmaa runner galaxy.jobs DEBUG 2015-03-03 10:06:25,943 (20175) Persisting job destination (destination id: sge) galaxy.jobs.handler INFO 2015-03-03 10:06:25,994 (20175) Job dispatched galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:26,584 (20175) submitting file /galaxy_data/job_working_directory/020/20175/galaxy_20175.sh galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:26,584 (20175) command is: samtools sort -n /galaxy_data/files/028/dataset_28339.dat /galaxy_data/files/028/dataset_28631.dat ; mv /galaxy_data/files/028/dataset_28631.dat.bam /galaxy_data/files/028/dataset_28631.dat; return_code=$?; cd /data12/galaxy/galaxy-env/galaxy; /data12/galaxy/galaxy-env/galaxy/set_metadata.sh /galaxy_data/files /galaxy_data/job_working_directory/020/20175 . /data12/galaxy/galaxy-env/galaxy/universe_wsgi.ini /galaxy_data/tmp/tmp28hp1G /galaxy_data/job_working_directory/020/20175/galaxy.json /galaxy_data/job_working_directory/020/20175/metadata_in_HistoryDatasetAssociation_35900_vmNjk7,/galaxy_data/job_working_directory/020/20175/metadata_kwds_HistoryDatasetAssociation_35900_IHm6c9,/galaxy_data/job_working_directory/020/20175/metadata_out_HistoryDatasetAssociation_35900_CboGKT,/galaxy_data/job_working_directory/020/20175/metadata_results_HistoryDatasetAssociation_35900_PbjjDh,,/galaxy_data/job_working_directory/020/20175/metadata_override_HistoryDatasetAssociation_35900_XD1a0c; sh -c "exit $return_code" galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:26,585 (20175) native specification is: -q galaxy galaxy.jobs.runners.drmaa INFO 2015-03-03 10:06:26,818 (20175) queued as 160573 galaxy.jobs DEBUG 2015-03-03 10:06:26,874 (20175) Persisting job destination (destination id: sge) galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:27,907 (20175/160573) state change: job is queued and active galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:06:35,538 (20175/160573) state change: job is running galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:12:10,200 (20173/160571) state change: job finished normally galaxy.datatypes.metadata DEBUG 2015-03-03 10:12:10,451 loading metadata from file for: HistoryDatasetAssociation 35898 galaxy.jobs DEBUG 2015-03-03 10:12:10,547 job 20173 ended galaxy.datatypes.metadata DEBUG 2015-03-03 10:12:10,551 Cleaning up external metadata files galaxy.jobs.runners.drmaa DEBUG 2015-03-03 10:46:16,161 (20175/160573) state change: job finished normally galaxy.jobs DEBUG 2015-03-03 10:46:16,371 setting dataset state to ERROR galaxy.jobs DEBUG 2015-03-03 10:46:16,464 job 20175 ended
I don't know why galaxy give the "setting dataset state to ERROR" line.
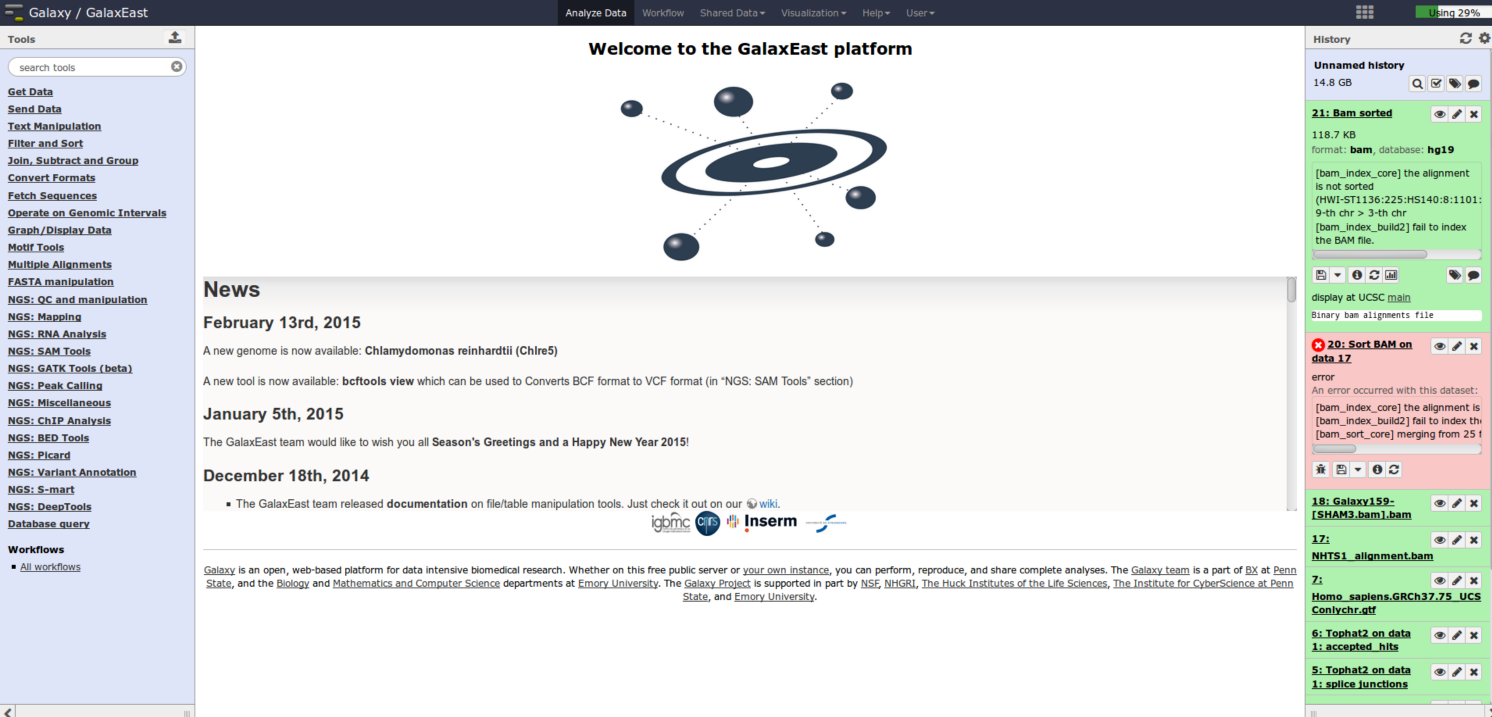
In attachment, you can see "21 : Bam sorted" on the test file and the "20 : Sort BAM on data 17" on the real file. You can see that it's not the "[bam_index_core] the alignment is not sorted (HISEQ:123:HBFN8ADXX:1:1101:1341:3979): 24-th chr > 13-th chr
[bam_index_build2] fail to index the BAM file." line which generate the error.
Thank you in advance for your help.
Have a nice day.
Best regards,
Amandine Velt Plateforme Biopuces et Séquençage +33388653529
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: https://lists.galaxyproject.org/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
participants (2)
-
 Amandine VELT
Amandine VELT -
 John Chilton
John Chilton