Could use some help with adding own perl scripts

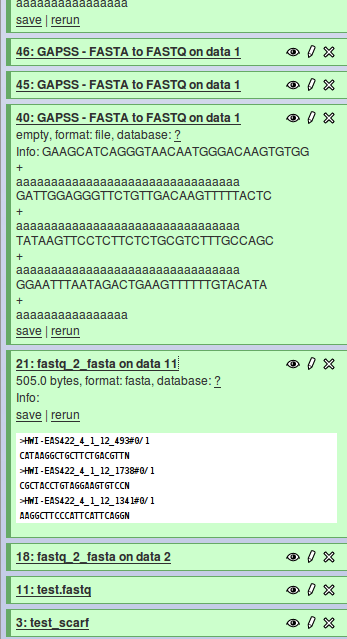
Hi, I recently started with galaxy, I got the : Galaxy build: $Rev 1733:a4214de3752e$ Now I need to add my own perl scipts to galaxy. This all goes fine, but there are some problems with the galaxy output look at the attachment: job 21 has an output in the white field, unlike job 40, I just can't seem to find out why? here are the xml's #######21############# <tool id="fast q 2 fasta " name="fastq_2_fasta"> <description>for each sequence in a file</description> <command interpreter="perl">fq2fa.pl $input $output</command> <inputs> <param format="fastq" name="input" type="data" label="Source file"/> </inputs> <outputs> <data format="fasta" name="output" /> </outputs> <tests> <test> <param name="input" value="fa_gc_content_input.fa"/> <output name="out_file1" file="fa_gc_content_output.txt"/> </test> </tests> <help> This tool computes GC content from a FASTA file. </help> </tool> ############################# ###########40################ <tool id="GAPSS_FASTA_to_FASTQ" name="GAPSS - FASTA to FASTQ"> <description>converter</description> <command interpreter="perl">GAPSS_FASTA2FASTQ.v2.pl $input $score $output</command> <inputs> <param format="fasta" name="input" type="data" label="FASTA File to convert" /> <param name="score" type="text" value="a" label="score" help="Use a if unsure." /> </inputs> <outputs> <data name="output" format="fastq" type="data" /> </outputs> <help> **What it does** This tool converts data from FASTQ format to FASTA format. #run as: perl GAPSS_FASTA2FASTQ.pl "FASTA file" "score to use or blank" #this script converts a FASTA file over to FASTQ #quality scores are by default Sanger, but can be adjusted #notes for user: # best sanger score = # best Solexa score = #input: FASTA file, and quality score to use #output: FASTQ file This tool is based on `GAPSS` by Matt and Michiel. http://www.lgtc.nl/GAPSS/ </help> </tool> ################ The perl script That needs to be added is for job 40 and looks like: ########PERl script job 40 ########### #!/usr/bin/perl #run as: perl GAPSS_FASTA2FASTQ.pl "FASTA file" "score to use or blank" #this script converts a FASTA file over to FASTQ #quality scores are by default Sanger, but can be adjusted #notes for user: # best sanger score = # best Solexa score = #input: FASTA file, and quality score to use #output: FASTQ file my $file_to_convert = $ARGV[0]; my $fastq_output_file_name = $file_to_convert.".fastq"; my $value_for_quality_score = $ARGV[1]; #set QC value to Sanger best () if no value given $value_for_quality_score =~ s/\n//; if ($value_for_quality_score eq ""){ $value_for_quality_score="a"; } #check is fasta format my $line1to4_fasta = `head -n 4 $file_to_convert`; my @array_line1to4_fasta=split(/\n/,$line1to4_fasta); if ( (@array_line1to4_fasta[0]=~/^>/) && (@array_line1to4_fasta[1]=~/[TAGCtagcNn]*/) && (@array_line1to4_fasta[2]=~/^>/) && (@array_line1to4_fasta[3]=~/[TAGCtagcNn]*/) ){}else{die "$file_to_convert is not a proper fasta file, terminated analysis\n";} #open file, open outfile, convert and print output, close infile, close outfile open(fasta_file,$file_to_convert)||die "could not open fasta file $file_to_convert\n"; open(fastq_file,">$fastq_output_file_name")||die "could not open fastq file $fastq_output_file_name to print to\n"; while (<fasta_file>){ chop; $_ =~ s/\n//; if ($_ =~ /^>/){ $_ =~ s/^>/@/;print fastq_file "$_\n"; }else{ print fastq_file "$_\n"; print fastq_file "+\n"; my $quality_line = $_; $quality_line =~ s/[TAGCtagcNn]/$value_for_quality_score/g; print fastq_file "$quality_line\n"; } } close (fasta_file)||die "could not close fasta file $file_to_convert \n"; close (fastq_file)||die "could not close fastq file $fastq_output_file_name\n"; ############################################ It just reformats a file and writes direct a file with .fastq behind it. I know there are other tools in galaxy but I just want mine to be put in and I just cant seem to get it to work. Can you give me some advise on the input and output parameters and how I control the white screen in galaxy, I just want the file that is written to be accessed by galaxy not the data displayed in the white screen, it will be for next generation sequencing and there wille be lots of fastq lines there. Thanks in advance. //Michel Villerius Human and Clinical Genetics Leiden University Medical Center
{kind=link}

Michel I am not sure, whether I do understand your e-mail correctly, but something looks strange to me: you call a perl script with 3 arguments :
###########40################ <tool id="GAPSS_FASTA_to_FASTQ" name="GAPSS - FASTA to FASTQ"> <description>converter</description> <command interpreter="perl">GAPSS_FASTA2FASTQ.v2.pl $input $score $output</command>
but you never use "$ARGV[2]" in your 'PERl script job 40' Have you tried my $fastq_output_file_name = $ARGV[2]; instead of " my $fastq_output_file_name = $file_to_convert.".fastq"; " Hans
Hi Hans, Thanks for the reply. I called the perl script with the 3rd argument because I don't understand the galaxy part of it, on command line I call it with just 2 arguments. How does the galaxy work with the output that is just a file ? What is the right way to handle the output with galaxy? In short: I can't even seem to get a command line working program working under galaxy without some proper manual, even google is not my best friend here when it comes to galaxy framework. //Michel On Tue, 2009-12-01 at 16:03 +0100, Hotz, Hans-Rudolf wrote:
Michel
I am not sure, whether I do understand your e-mail correctly, but something looks strange to me:
you call a perl script with 3 arguments :
###########40################ <tool id="GAPSS_FASTA_to_FASTQ" name="GAPSS - FASTA to FASTQ"> <description>converter</description> <command interpreter="perl">GAPSS_FASTA2FASTQ.v2.pl $input $score $output</command>
but you never use "$ARGV[2]" in your 'PERl script job 40'
Have you tried
my $fastq_output_file_name = $ARGV[2];
instead of " my $fastq_output_file_name = $file_to_convert.".fastq"; "
Hans

Hello Michel, have you seen our wiki for adding a new tool? http://bitbucket.org/galaxy/galaxy-central/wiki/AddToolTutorial Greg On Dec 1, 2009, at 10:19 AM, Michel P. Villerius wrote:
Hi Hans,
Thanks for the reply.
I called the perl script with the 3rd argument because I don't understand the galaxy part of it, on command line I call it with just 2 arguments. How does the galaxy work with the output that is just a file ? What is the right way to handle the output with galaxy? In short: I can't even seem to get a command line working program working under galaxy without some proper manual, even google is not my best friend here when it comes to galaxy framework.
//Michel
On Tue, 2009-12-01 at 16:03 +0100, Hotz, Hans-Rudolf wrote:
Michel
I am not sure, whether I do understand your e-mail correctly, but something looks strange to me:
you call a perl script with 3 arguments :
###########40################ <tool id="GAPSS_FASTA_to_FASTQ" name="GAPSS - FASTA to FASTQ"> <description>converter</description> <command interpreter="perl">GAPSS_FASTA2FASTQ.v2.pl $input $score $output</command>
but you never use "$ARGV[2]" in your 'PERl script job 40'
Have you tried
my $fastq_output_file_name = $ARGV[2];
instead of " my $fastq_output_file_name = $file_to_convert.".fastq"; "
Hans
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Greg Von Kuster Galaxy Development Team greg@bx.psu.edu
Thanks Greg, It was my starting point. I now know that the best way to go is to write a wrapper script for my tools to use in galaxy. //Michel On Tue, 2009-12-01 at 11:12 -0500, Greg Von Kuster wrote:
Hello Michel,
have you seen our wiki for adding a new tool?
http://bitbucket.org/galaxy/galaxy-central/wiki/AddToolTutorial
Greg
On Dec 1, 2009, at 10:19 AM, Michel P. Villerius wrote:
Hi Hans,
Thanks for the reply.
I called the perl script with the 3rd argument because I don't understand the galaxy part of it, on command line I call it with just 2 arguments. How does the galaxy work with the output that is just a file ? What is the right way to handle the output with galaxy? In short: I can't even seem to get a command line working program working under galaxy without some proper manual, even google is not my best friend here when it comes to galaxy framework.
//Michel
On Tue, 2009-12-01 at 16:03 +0100, Hotz, Hans-Rudolf wrote:
Michel
I am not sure, whether I do understand your e-mail correctly, but something looks strange to me:
you call a perl script with 3 arguments :
###########40################ <tool id="GAPSS_FASTA_to_FASTQ" name="GAPSS - FASTA to FASTQ"> <description>converter</description> <command interpreter="perl">GAPSS_FASTA2FASTQ.v2.pl $input $score $output</command>
but you never use "$ARGV[2]" in your 'PERl script job 40'
Have you tried
my $fastq_output_file_name = $ARGV[2];
instead of " my $fastq_output_file_name = $file_to_convert.".fastq"; "
Hans
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Greg Von Kuster Galaxy Development Team greg@bx.psu.edu
I'm almost done with the wrapper scripts, galaxy works great, still there is one question left: One of my perl script creates one or more output files, I wrote a wrapper that handles one file within galaxy and that works great. How do I specify two or more output files with galaxy if I on forehand don't know how many files there will be created? Can I use an array in the wrapper perl script(@) that returns the array to galaxy? Thanks, //Michel On Tue, 2009-12-01 at 11:12 -0500, Greg Von Kuster wrote:
Hello Michel,
have you seen our wiki for adding a new tool?
http://bitbucket.org/galaxy/galaxy-central/wiki/AddToolTutorial
Greg
On Dec 1, 2009, at 10:19 AM, Michel P. Villerius wrote:
Hi Hans,
Thanks for the reply.
I called the perl script with the 3rd argument because I don't understand the galaxy part of it, on command line I call it with just 2 arguments. How does the galaxy work with the output that is just a file ? What is the right way to handle the output with galaxy? In short: I can't even seem to get a command line working program working under galaxy without some proper manual, even google is not my best friend here when it comes to galaxy framework.
//Michel
On Tue, 2009-12-01 at 16:03 +0100, Hotz, Hans-Rudolf wrote:
Michel
I am not sure, whether I do understand your e-mail correctly, but something looks strange to me:
you call a perl script with 3 arguments :
###########40################ <tool id="GAPSS_FASTA_to_FASTQ" name="GAPSS - FASTA to FASTQ"> <description>converter</description> <command interpreter="perl">GAPSS_FASTA2FASTQ.v2.pl $input $score $output</command>
but you never use "$ARGV[2]" in your 'PERl script job 40'
Have you tried
my $fastq_output_file_name = $ARGV[2];
instead of " my $fastq_output_file_name = $file_to_convert.".fastq"; "
Hans
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Greg Von Kuster Galaxy Development Team greg@bx.psu.edu
Hello Michel, Any number of output files can be generated by naming them in a certain way and putting them in a certain directory. The maf_to_interval tool in ~/tools/maf/maf_to_interval.xml and maf_to_interval.py is an example of how this can be done. On Dec 3, 2009, at 7:43 AM, Michel P. Villerius wrote:
I'm almost done with the wrapper scripts, galaxy works great, still there is one question left:
One of my perl script creates one or more output files, I wrote a wrapper that handles one file within galaxy and that works great. How do I specify two or more output files with galaxy if I on forehand don't know how many files there will be created? Can I use an array in the wrapper perl script(@) that returns the array to galaxy?
Thanks,
//Michel
On Tue, 2009-12-01 at 11:12 -0500, Greg Von Kuster wrote:
Hello Michel,
have you seen our wiki for adding a new tool?
http://bitbucket.org/galaxy/galaxy-central/wiki/AddToolTutorial
Greg
On Dec 1, 2009, at 10:19 AM, Michel P. Villerius wrote:
Hi Hans,
Thanks for the reply.
I called the perl script with the 3rd argument because I don't understand the galaxy part of it, on command line I call it with just 2 arguments. How does the galaxy work with the output that is just a file ? What is the right way to handle the output with galaxy? In short: I can't even seem to get a command line working program working under galaxy without some proper manual, even google is not my best friend here when it comes to galaxy framework.
//Michel
On Tue, 2009-12-01 at 16:03 +0100, Hotz, Hans-Rudolf wrote:
Michel
I am not sure, whether I do understand your e-mail correctly, but something looks strange to me:
you call a perl script with 3 arguments :
###########40################ <tool id="GAPSS_FASTA_to_FASTQ" name="GAPSS - FASTA to FASTQ"> <description>converter</description> <command interpreter="perl">GAPSS_FASTA2FASTQ.v2.pl $input $score $output</command>
but you never use "$ARGV[2]" in your 'PERl script job 40'
Have you tried
my $fastq_output_file_name = $ARGV[2];
instead of " my $fastq_output_file_name = $file_to_convert.".fastq"; "
Hans
_______________________________________________ galaxy-dev mailing list galaxy-dev@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-dev
Greg Von Kuster Galaxy Development Team greg@bx.psu.edu
Greg Von Kuster Galaxy Development Team greg@bx.psu.edu
participants (3)
-
 Greg Von Kuster
Greg Von Kuster -
 Hotz, Hans-Rudolf
Hotz, Hans-Rudolf -
 Michel P. Villerius
Michel P. Villerius