
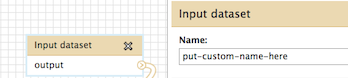
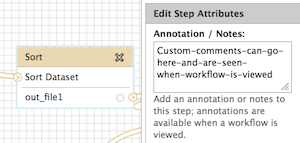
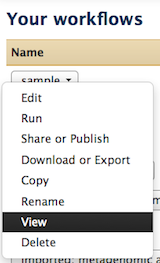
Hi Jun, The name of the tool itself cannot be modified, but there are workflow annotations that can help to communicate usage instructions to those you share it with. Specifically, 1) where each specific input should be selected and 2) what is happening at each step. For 1: where each specific input should be selected Unique to input datasets, the "name" field can be used. This will be displayed when a workflow is run, underneath the tool name, but above the pull down menu of datasets (for that step). Try a custom name here, save, then click run (all from the workflow menu) to see how the display works. In the workflow editor, this is where the input "name" attribute is located: For 2: what is happening at each step There is an annotation/notes box for all tools (including the input tool) where more help or comments can be entered. This is not displayed at run time, but when a workflow's structure is "viewed". You can find examples of workflows with this sort of annotation in the "Shared Data -> Published Workflows" area, or test out and see if you find it helpful. This is where it is located in the workflow editor: How to "view" a workflow: Click on a workflow's button on just about any Galaxy web page that it is displayed on. For example, "Shared Data -> Published Workflows", click on the workflow at the top of the list. Right now it is "metagenomic analysis" and this one has annotation (the next two do as well). When at "Your workflows" or "Workflows shared with you by others", click on the down arrow to the right side of the workflow name within the button, and "View" will be one of the options. I realize this is not exactly what you wanted originally, but hopefully these options will help you to communicate what you need to, Jen Galaxy team On 8/21/13 5:22 PM, Jun Fan wrote:
Hi Jen,
Maybe I have not explained my question cleared which caused the confusion: I did not mean the name of dataset. Actually I asked about the display of the tool in the graphic view from the default value (guess it is the name attribute in the tool element of the wrapper) to something else in the workflow editor. As my ultimate purpose is to share my workflow with someone else. If they see three steps with the same displayed name and do not have the related knowledge, they will get lost. To help myself to explain well, here is the illustration:
From
------------------------------- | mzidLib:PostProcessing| | mzidLib:PostProcessing| | mzidLib:PostProcessing| |-----------------------------| |-----------------------------| |-----------------------------| | input file | ---------->| input file | ---------->| input file | |-----------------------------| | |-----------------------------| | |-----------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ------------------------------- |-----------------------------| |-----------------------------| To ------------------------------------- ------------------------------------------- ------------------------------------------------ | mzidLib:PostProcessing FDR| | mzidLib:PostProcessing Threshold| | mzidLib:PostProcessing ProteoGroup| |----------------------------------| |------------------------------------------| |----------------------------------------------| | input file | ---------->| input file | ------------>| input file | |----------------------------------| | |------------------------------------------| | |----------------------------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ----------------------------------- |------------------------------------------| |----------------------------------------------|
Best regards! Jun
-- Jennifer Hillman-Jackson http://galaxyproject.org
{kind=link}
{kind=link}
{kind=link}