
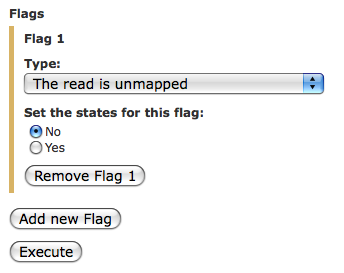
Michaela: Sorry for delay in replying. Looks like you need to do this: 1. Map reads with bowtie using "try hard" option. 2. Leave -m option at -1 3. Set -k ridiculously high (e.g., 1000) 4. Once mapped filter SAM with "SAM Tools->Filter SAM on bitwise flag" filter tool using option as in the attached image 5. Convert SAM format to intervals using "Covert SAM to interval" tool. 6. Subtract coordinates of regions from the previous step from coordinates of your exons using "Operate on Genome Intervals-
Subtract" tool. The result will contain unmapped regions.
Let me know how it goes, and always cope to galaxy-user mailing list. Thanks, anton galaxy team On Jan 20, 2010, at 2:16 PM, Michaela Lee wrote:
Hi Anton,
I am sorry to keep bothering you but could you clarify a bit more on what I can do with mapped reads on Galaxy? Here is my situation: We have attempted to sequence all the exons on the X chromosome for some of our subjects through exon capture and high through-put sequencing. We have all the Illumina reads on an external hard drive and would like to know which regions of our reference sequence were not "captured" or sequenced after mapping all the reads using Maq or Bowtie. We are not interested in unmapped reads, just the unmapped regions of the reference sequence.
I have watched video #7 of Galaxy but I am confused as to which "bitwise flag" I would use to have Galaxy generate an output file with a list of regions of the reference sequence that were not mapped. Ultimately, we would like to compare all the regions that were not captured for all our subjects to see if those unsequenced regions are random or not.
I appreciate any help you can give me. Thank you!
Michaela
Anton Nekrutenko http://nekrut.bx.psu.edu http://usegalaxy.org
{kind=link}