
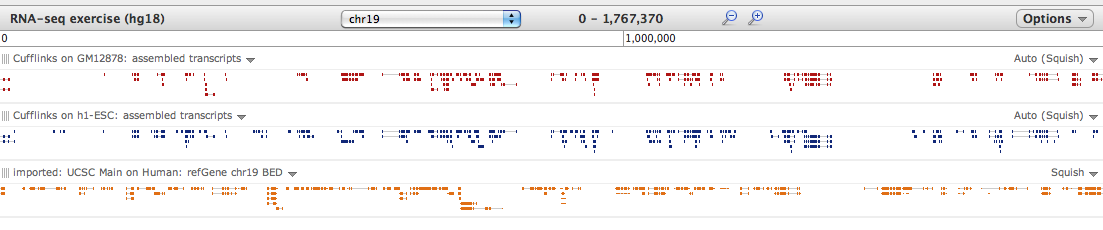
Hello Aleks, Thank you for bring these hg18/hg19 problem to our attention - it was introduced during an edit last week and has now been corrected. The good news is that results are obtained with either - the issue with overlap may be with the Visualization tool itself. When very "zoomed out", the overlap regions can vanish if small enough (we will be correcting/normalizing for this). However, the data is there if you just zoom in, as in the attached screenshot. Hopefully this helps! Best, Jen Galaxy team On 6/24/11 6:19 AM, Aleks Schein wrote:
Dear all,
I tried to follow the Cufflinks tutorial created by Jeremy. The problems start with first "visualization" step. The is no overlapping at all between Cufflinks transcripts and RefSeq Chr19 reference, provided on the tutorial page. Was that the goal, or transcripts should overlap and something did not work? By the way, mapping for cufflinks was done to hg19, but reference annotation is provided at hg18. Maybe, this is the problem?
Aleks
Quoting Robert Curtis Hendrickson <curtish@uab.edu>:
Jen,
We are running into the same problem on our local install of galaxy. We're running Cufflinks v.1.0.1, on a BAM file (accepted_reads) from TopHat run on mm9 based RNAseq data (paired-end 25mer), and pulled down the changes made to galaxy last month to support the 1.0.1 version of Cufflinks.
We (think) we have mm9 indexes locally installed. We can successfully run get_genomic_sequence on mm9 .BED's....
Turning off bias correction made no difference.
We also tried rolling back to Cufflinks v0.9.1 (including the Galaxy patch), and got the same error
An error occurred running this job: cufflinks v0.9.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
An error occurred running this job: cufflinks v1.0.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
I can provide a link to the history on our server that you should (theoretically) be able to access.
Regards, Curtis
-----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user- bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: Friday, June 10, 2011 3:58 PM To: David Robinson Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] Error running cufflinks on Galaxy
Hello David,
Cufflinks requires locally cached data to perform the Bias Correction function.
Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem.
Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible).
Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine).
Jen Galaxy team
On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get an error:
An error occurred running this job: /cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from- uploaded-file
Thanks in advance for your help!
-David
************************************************
David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630
************************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org/ http://galaxyproject.org/
{kind=link}