Re: [galaxy-user] galaxy-user Digest, Vol 86, Issue 17

Hi Jen, Maybe I have not explained my question cleared which caused the confusion: I did not mean the name of dataset. Actually I asked about the display of the tool in the graphic view from the default value (guess it is the name attribute in the tool element of the wrapper) to something else in the workflow editor. As my ultimate purpose is to share my workflow with someone else. If they see three steps with the same displayed name and do not have the related knowledge, they will get lost. To help myself to explain well, here is the illustration:
From
------------------------------- | mzidLib:PostProcessing| | mzidLib:PostProcessing| | mzidLib:PostProcessing| |-----------------------------| |-----------------------------| |-----------------------------| | input file | ---------->| input file | ---------->| input file | |-----------------------------| | |-----------------------------| | |-----------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ------------------------------- |-----------------------------| |-----------------------------| To ------------------------------------- ------------------------------------------- ------------------------------------------------ | mzidLib:PostProcessing FDR| | mzidLib:PostProcessing Threshold| | mzidLib:PostProcessing ProteoGroup| |----------------------------------| |------------------------------------------| |----------------------------------------------| | input file | ---------->| input file | ------------>| input file | |----------------------------------| | |------------------------------------------| | |----------------------------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ----------------------------------- |------------------------------------------| |----------------------------------------------| Best regards! Jun Date: Tue, 20 Aug 2013 11:34:47 -0700 From: Jennifer Jackson <jen@bx.psu.edu> To: Jun Fan <j.fan@qmul.ac.uk> Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] customize tool display in the workflow Message-ID: <5213B6C7.5030800@bx.psu.edu> Content-Type: text/plain; charset="iso-8859-1"; Format="flowed" Hi Jun, You are very close - just click on the "create" button within the "Edit Step Actions" box and it will expand, where you can then enter the new custom name. It will look something like this: The other post you are referring to is a method to name the output dataset based on the input datasets name. You can certainly try this out and see if it is useful. Hope this helps, Jen Galaxy team On 8/20/13 9:34 AM, Jun Fan wrote:
Hi Jennifer,
Many thanks for your reply. Unfortunately I am not clever to figure out how to using the existing workflow dataset renaming functions. I only know how to rename a dataset within history. Could you show me how to do this? I have attached a screen shot of my workflow. You can see that I have only managed to add annotation/notes to this step. Ideally I would like to have the step with the label of "mzidLib:PostProcessing FDR" in the graphic view. From other thread, someone mentioned to use ${method} in the label, I failed to apply this trick.
Best regards! Jun
Message: 3 Date: Mon, 19 Aug 2013 10:18:32 -0700 From: Jennifer Jackson <jen@bx.psu.edu> To: Jun Fan <j.fan@qmul.ac.uk> Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] customize tool display in the workflow Message-ID: <52125368.1060308@bx.psu.edu> Content-Type: text/plain; charset="iso-8859-1"; Format="flowed"
Hi Jun,
I asked Dannon (our workflow lead developer) and he suggested just using the existing workflow datastet renaming functions. These are in the right panel when you click on a dataset within the workflow editor (as you probably know).
Inherited naming is not something that is currently being worked on, but you can always start a Trello card and see if it gathers votes or the attention of a contributor from the larger development community: http://wiki.galaxyproject.org/Support#Galaxy_Issue_Board
Take care,
Jen Galaxy team
On 8/18/13 5:11 PM, Jun Fan wrote:
Dear Galaxy develop team:
Normally I generate the workflow from history. It works fine except one annoying problem: the display of step/tool is just the tool name, which can cause confusion in a big workflow. For example, I have a mixture of samples from multiple species and do the BLAST against Human, Virus, Mouse etc which will call BLAST several times. To make results distinguishable, I will rename the dataset in the history. But this will be lost when creating workflow from history. The workflow will have duplicate steps named as BLAST while I really want to have steps displayed as BLAST against Human etc. Could you tell me whether I have done something wrong? If not, could you kindly add this functionality into the new release of Galaxy? If it is possible to customize the tool display in the workflow, how to write the wrapper as setting the label to allow this?
Best regards!
Jun
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
http://galaxyproject.org/search/mailinglists/ -- Jennifer Hillman-Jackson http://galaxyproject.org
-------------- next part -------------- An HTML attachment was scrubbed... URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20130819/8d ca342d /attachment-0001.html>
------------------------------
_______________________________________________ galaxy-user mailing list galaxy-user@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-user
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
End of galaxy-user Digest, Vol 86, Issue 16 *******************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
-- Jennifer Hillman-Jackson http://galaxyproject.org

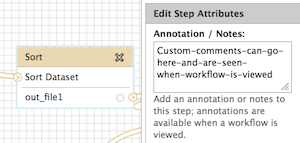
Hi Jun, Thanks for the clarification. Workflow step annotations would best suit your needs, I think. When you're in the editor, click on any step and on the right side you'll see a field "Annotation / Notes" which will be displayed when the workflow is viewed. This can be used to clear up any confusion about what the individual step does. Good luck! Dannon On Wed, Aug 21, 2013 at 8:22 PM, Jun Fan <j.fan@qmul.ac.uk> wrote:
Hi Jen,
Maybe I have not explained my question cleared which caused the confusion: I did not mean the name of dataset. Actually I asked about the display of the tool in the graphic view from the default value (guess it is the name attribute in the tool element of the wrapper) to something else in the workflow editor. As my ultimate purpose is to share my workflow with someone else. If they see three steps with the same displayed name and do not have the related knowledge, they will get lost. To help myself to explain well, here is the illustration:
From
------------------------------- | mzidLib:PostProcessing| | mzidLib:PostProcessing| | mzidLib:PostProcessing| |-----------------------------| |-----------------------------| |-----------------------------| | input file | ---------->| input file | ---------->| input file | |-----------------------------| | |-----------------------------| | |-----------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ------------------------------- |-----------------------------| |-----------------------------| To ------------------------------------- ------------------------------------------- ------------------------------------------------ | mzidLib:PostProcessing FDR| | mzidLib:PostProcessing Threshold| | mzidLib:PostProcessing ProteoGroup| |----------------------------------| |------------------------------------------| |----------------------------------------------| | input file | ---------->| input file | ------------>| input file | |----------------------------------| | |------------------------------------------| | |----------------------------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ----------------------------------- |------------------------------------------| |----------------------------------------------|
Best regards! Jun
Date: Tue, 20 Aug 2013 11:34:47 -0700 From: Jennifer Jackson <jen@bx.psu.edu> To: Jun Fan <j.fan@qmul.ac.uk> Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] customize tool display in the workflow Message-ID: <5213B6C7.5030800@bx.psu.edu> Content-Type: text/plain; charset="iso-8859-1"; Format="flowed"
Hi Jun,
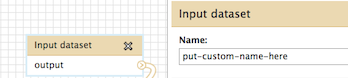
You are very close - just click on the "create" button within the "Edit Step Actions" box and it will expand, where you can then enter the new custom name. It will look something like this:
The other post you are referring to is a method to name the output dataset based on the input datasets name. You can certainly try this out and see if it is useful.
Hope this helps,
Jen Galaxy team
On 8/20/13 9:34 AM, Jun Fan wrote:
Hi Jennifer,
Many thanks for your reply. Unfortunately I am not clever to figure out how to using the existing workflow dataset renaming functions. I only know how to rename a dataset within history. Could you show me how to do this? I have attached a screen shot of my workflow. You can see that I have only managed to add annotation/notes to this step. Ideally I would like to have the step with the label of "mzidLib:PostProcessing FDR" in the graphic view. From other thread, someone mentioned to use ${method} in the label, I failed to apply this trick.
Best regards! Jun
Message: 3 Date: Mon, 19 Aug 2013 10:18:32 -0700 From: Jennifer Jackson <jen@bx.psu.edu> To: Jun Fan <j.fan@qmul.ac.uk> Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] customize tool display in the workflow Message-ID: <52125368.1060308@bx.psu.edu> Content-Type: text/plain; charset="iso-8859-1"; Format="flowed"
Hi Jun,
I asked Dannon (our workflow lead developer) and he suggested just using the existing workflow datastet renaming functions. These are in the right panel when you click on a dataset within the workflow editor (as you probably know).
Inherited naming is not something that is currently being worked on, but you can always start a Trello card and see if it gathers votes or the attention of a contributor from the larger development community: http://wiki.galaxyproject.org/Support#Galaxy_Issue_Board
Take care,
Jen Galaxy team
On 8/18/13 5:11 PM, Jun Fan wrote:
Dear Galaxy develop team:
Normally I generate the workflow from history. It works fine except one annoying problem: the display of step/tool is just the tool name, which can cause confusion in a big workflow. For example, I have a mixture of samples from multiple species and do the BLAST against Human, Virus, Mouse etc which will call BLAST several times. To make results distinguishable, I will rename the dataset in the history. But this will be lost when creating workflow from history. The workflow will have duplicate steps named as BLAST while I really want to have steps displayed as BLAST against Human etc. Could you tell me whether I have done something wrong? If not, could you kindly add this functionality into the new release of Galaxy? If it is possible to customize the tool display in the workflow, how to write the wrapper as setting the label to allow this?
Best regards!
Jun
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
http://galaxyproject.org/search/mailinglists/ -- Jennifer Hillman-Jackson http://galaxyproject.org
-------------- next part -------------- An HTML attachment was scrubbed... URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20130819/8d ca342d /attachment-0001.html>
------------------------------
_______________________________________________ galaxy-user mailing list galaxy-user@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-user
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
End of galaxy-user Digest, Vol 86, Issue 16 *******************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
-- Jennifer Hillman-Jackson http://galaxyproject.org

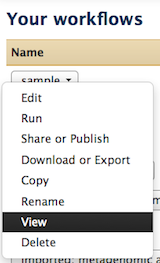
Hi Jun, The name of the tool itself cannot be modified, but there are workflow annotations that can help to communicate usage instructions to those you share it with. Specifically, 1) where each specific input should be selected and 2) what is happening at each step. For 1: where each specific input should be selected Unique to input datasets, the "name" field can be used. This will be displayed when a workflow is run, underneath the tool name, but above the pull down menu of datasets (for that step). Try a custom name here, save, then click run (all from the workflow menu) to see how the display works. In the workflow editor, this is where the input "name" attribute is located: For 2: what is happening at each step There is an annotation/notes box for all tools (including the input tool) where more help or comments can be entered. This is not displayed at run time, but when a workflow's structure is "viewed". You can find examples of workflows with this sort of annotation in the "Shared Data -> Published Workflows" area, or test out and see if you find it helpful. This is where it is located in the workflow editor: How to "view" a workflow: Click on a workflow's button on just about any Galaxy web page that it is displayed on. For example, "Shared Data -> Published Workflows", click on the workflow at the top of the list. Right now it is "metagenomic analysis" and this one has annotation (the next two do as well). When at "Your workflows" or "Workflows shared with you by others", click on the down arrow to the right side of the workflow name within the button, and "View" will be one of the options. I realize this is not exactly what you wanted originally, but hopefully these options will help you to communicate what you need to, Jen Galaxy team On 8/21/13 5:22 PM, Jun Fan wrote:
Hi Jen,
Maybe I have not explained my question cleared which caused the confusion: I did not mean the name of dataset. Actually I asked about the display of the tool in the graphic view from the default value (guess it is the name attribute in the tool element of the wrapper) to something else in the workflow editor. As my ultimate purpose is to share my workflow with someone else. If they see three steps with the same displayed name and do not have the related knowledge, they will get lost. To help myself to explain well, here is the illustration:
From
------------------------------- | mzidLib:PostProcessing| | mzidLib:PostProcessing| | mzidLib:PostProcessing| |-----------------------------| |-----------------------------| |-----------------------------| | input file | ---------->| input file | ---------->| input file | |-----------------------------| | |-----------------------------| | |-----------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ------------------------------- |-----------------------------| |-----------------------------| To ------------------------------------- ------------------------------------------- ------------------------------------------------ | mzidLib:PostProcessing FDR| | mzidLib:PostProcessing Threshold| | mzidLib:PostProcessing ProteoGroup| |----------------------------------| |------------------------------------------| |----------------------------------------------| | input file | ---------->| input file | ------------>| input file | |----------------------------------| | |------------------------------------------| | |----------------------------------------------| |output (mzid) |----- |output (mzid) |----- |output (mzid) | ----------------------------------- |------------------------------------------| |----------------------------------------------|
Best regards! Jun
-- Jennifer Hillman-Jackson http://galaxyproject.org
{kind=link}
{kind=link}
{kind=link}
participants (3)
-
 Dannon Baker
Dannon Baker -
 Jennifer Jackson
Jennifer Jackson -
 Jun Fan
Jun Fan