FASTQ splitter empty and FASTQ manipulator doesn't work

I am having the same issue as this user: http://user.list.galaxyproject.org/FASTQ-splitter-produced-empty-dataset-ple... I have a fastq file, in which both paired end reads were concatenated into a single file. @HWI-ST632:288:C2L0BACXX:8:1101:1639:1746 1:N:0:GCCAAT NTTTGTCTCTGGTCTGTACTTGTGGGCCAGCTTAAGCAGCTGAGTAGCTGTTTGGCGGTCCAGGGCCTGGGTGAACTGGTTAATCGCAGGAGGCACTTTCA + #1=DDFFFHHHGFHJIHJIJJIHHJJJIIIIIJJGIGIGIJIGIDGHJIIJJJJJIJJGHHGFFFFDDDDDCBBDCCDDCDDDEDDDDDDBDDDDBDDCCC @HWI-ST632:288:C2L0BACXX:8:1101:1876:1741 2:N:0:GCCAAT NTTTATTTTTCGTTATTGTTGGTGGTTTAAAAAATTCCCCCCATGTAATTATTGTGAACACCTTGCTTTGTGGTCACTGTAACATTTGGGGGGCGGGACAG Fastq splitter results in empty data sets because, as explained in the thread, the data is already split into their corresponding forward and reverse reads and splitter is meant to split reads in which the paired end reads have been joined tail to head. I tried the manipulate FASTQ tool as the thread above suggests, however the FASTQ sequence identifier is slightly different so the suggested Match of .1 and .2 example shown in the thread doesn't work, because my FASTQ file has the identifier in Casava 1.8 format, in which the identifier line does not end with the 1 or 2 for forward and reverse strand. Instead the strand is identified as the 1 or 2 after the space in the identifier line, so <space>1:N:0 . . . and <space>2:N:0 . . . . I tried running FASTQ manipulator with the Match Reads according to regular expression with " 1:" and " 2:" but that did not work, I ended up with two identical fastq files with same sequences as the original file, so it basically just made a copy and did not deconcatenate the forward or reverse. Am I using this Match Reads by regular expression wrong? Janice Patterson patterja@ohsu.edu

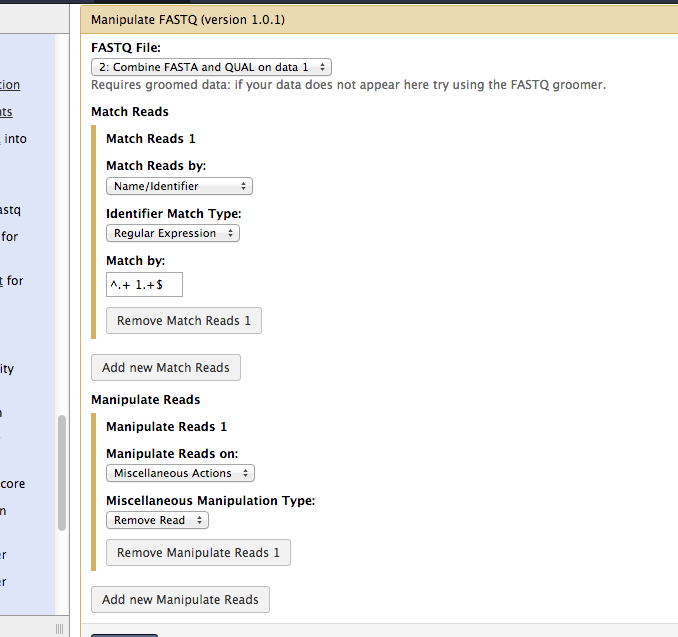
Hello Janice, The tool "Manipulate FASTQ" reads can be used on your data as well. It just needs a different regular expression for the sequence identifier match. A created two that make use of the internal space in the identifier to test for the presence of a "1" or a "2" to do the split: ^.+ 1.+$ ^.+ 2.+$ Run the tool twice. The match expression used in any run will be the sequences that are _/removed/_. Below is a graphic of the settings for an example run. Good luck! Jen Galaxy team On 1/4/14 3:02 PM, Janice Patterson wrote:
I am having the same issue as this user:
http://user.list.galaxyproject.org/FASTQ-splitter-produced-empty-dataset-ple...
I have a fastq file, in which both paired end reads were concatenated into a single file.
@HWI-ST632:288:C2L0BACXX:8:1101:1639:1746 1:N:0:GCCAAT NTTTGTCTCTGGTCTGTACTTGTGGGCCAGCTTAAGCAGCTGAGTAGCTGTTTGGCGGTCCAGGGCCTGGGTGAACTGGTTAATCGCAGGAGGCACTTTCA + #1=DDFFFHHHGFHJIHJIJJIHHJJJIIIIIJJGIGIGIJIGIDGHJIIJJJJJIJJGHHGFFFFDDDDDCBBDCCDDCDDDEDDDDDDBDDDDBDDCCC @HWI-ST632:288:C2L0BACXX:8:1101:1876:1741 2:N:0:GCCAAT NTTTATTTTTCGTTATTGTTGGTGGTTTAAAAAATTCCCCCCATGTAATTATTGTGAACACCTTGCTTTGTGGTCACTGTAACATTTGGGGGGCGGGACAG
Fastq splitter results in empty data sets because, as explained in the thread, the data is already split into their corresponding forward and reverse reads and splitter is meant to split reads in which the paired end reads have been joined tail to head.
I tried the manipulate FASTQ tool as the thread above suggests, however the FASTQ sequence identifier is slightly different so the suggested Match of .1 and .2 example shown in the thread doesn't work, because my FASTQ file has the identifier in Casava 1.8 format, in which the identifier line does not end with the 1 or 2 for forward and reverse strand. Instead the strand is identified as the 1 or 2 after the space in the identifier line, so <space>1:N:0 . . . and <space>2:N:0 . . . . I tried running FASTQ manipulator with the Match Reads according to regular expression with " 1:" and " 2:" but that did not work, I ended up with two identical fastq files with same sequences as the original file, so it basically just made a copy and did not deconcatenate the forward or reverse. Am I using this Match Reads by regular expression wrong?
Janice Patterson patterja@ohsu.edu
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
-- Jennifer Hillman-Jackson http://galaxyproject.org
{kind=link}
participants (2)
-
 Janice Patterson
Janice Patterson -
 Jennifer Jackson
Jennifer Jackson