Error running cufflinks on Galaxy

Hello, When I attempt to run cufflinks based on .sam output from bowtie I get an error: An error occurred running this job: *cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf' *What can I do to get around this problem and run cufflinks? My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file): http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from-upload... Thanks in advance for your help! -David ************************************************ David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630 ************************************************

Hello David, Cufflinks requires locally cached data to perform the Bias Correction function. Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem. Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible). Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine). Jen Galaxy team On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get an error:
An error occurred running this job: /cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from-upload...
Thanks in advance for your help!
-David
************************************************
David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630
************************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org http://galaxyproject.org

Hello, Dr. Jeremy Goecks already sent me an response that fixed the problem (perhaps forgetting to email the list as well)- I was using unsorted output directly from Bowtie. When I tried using a TopHat BAM file it worked. Thank you for your reply! -David On Jun 10, 2011 4:58pm, Jennifer Jackson <jen@bx.psu.edu> wrote:
Hello David,
Cufflinks requires locally cached data to perform the Bias Correction function.
Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem.
Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible).
Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine).
Jen
Galaxy team
On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get
an error:
An error occurred running this job: /cufflinks v1.0.1
cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b
/galaxy/data/hg19/sam_index/hg19.fa
Error running cufflinks. [Errno 2] No such file or directory:
'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran
it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from-upload...
Thanks in advance for your help!
-David
************************************************
David Robinson
Graduate Student
Lewis-Sigler Institute for Integrative Genomics
Carl Icahn Laboratory
Princeton University
646-620-6630
************************************************
___________________________________________________________
The Galaxy User list should be used for the discussion of
Galaxy analysis and other features on the public server
at usegalaxy.org. Please keep all replies on the list by
using "reply all" in your mail client. For discussion of
local Galaxy instances and the Galaxy source code, please
use the Galaxy Development list:
To manage your subscriptions to this and other Galaxy lists,
please use the interface at:
--
Jennifer Jackson

Jen, We are running into the same problem on our local install of galaxy. We're running Cufflinks v.1.0.1, on a BAM file (accepted_reads) from TopHat run on mm9 based RNAseq data (paired-end 25mer), and pulled down the changes made to galaxy last month to support the 1.0.1 version of Cufflinks. We (think) we have mm9 indexes locally installed. We can successfully run get_genomic_sequence on mm9 .BED's.... Turning off bias correction made no difference. We also tried rolling back to Cufflinks v0.9.1 (including the Galaxy patch), and got the same error An error occurred running this job: cufflinks v0.9.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf' An error occurred running this job: cufflinks v1.0.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf' I can provide a link to the history on our server that you should (theoretically) be able to access. Regards, Curtis
-----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user- bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: Friday, June 10, 2011 3:58 PM To: David Robinson Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] Error running cufflinks on Galaxy
Hello David,
Cufflinks requires locally cached data to perform the Bias Correction function.
Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem.
Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible).
Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine).
Jen Galaxy team
On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get an error:
An error occurred running this job: /cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from- uploaded-file
Thanks in advance for your help!
-David
************************************************
David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630
************************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Hello Robert, This guess turned out to be incorrect. The problem for this user was solved by either sorting the Bowtie output before using CuffLinks or by using TopHat output instead. Hopefully one of these will solve your issues as well, Best, Jen Galaxy team On 6/22/11 1:17 PM, Robert Curtis Hendrickson wrote:
Jen,
We are running into the same problem on our local install of galaxy. We're running Cufflinks v.1.0.1, on a BAM file (accepted_reads) from TopHat run on mm9 based RNAseq data (paired-end 25mer), and pulled down the changes made to galaxy last month to support the 1.0.1 version of Cufflinks.
We (think) we have mm9 indexes locally installed. We can successfully run get_genomic_sequence on mm9 .BED's....
Turning off bias correction made no difference.
We also tried rolling back to Cufflinks v0.9.1 (including the Galaxy patch), and got the same error
An error occurred running this job: cufflinks v0.9.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
An error occurred running this job: cufflinks v1.0.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
I can provide a link to the history on our server that you should (theoretically) be able to access.
Regards, Curtis
-----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user- bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: Friday, June 10, 2011 3:58 PM To: David Robinson Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] Error running cufflinks on Galaxy
Hello David,
Cufflinks requires locally cached data to perform the Bias Correction function.
Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem.
Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible).
Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine).
Jen Galaxy team
On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get an error:
An error occurred running this job: /cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from- uploaded-file
Thanks in advance for your help!
-David
************************************************
David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630
************************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org/ http://galaxyproject.org/

Dear all, I tried to follow the Cufflinks tutorial created by Jeremy. The problems start with first "visualization" step. The is no overlapping at all between Cufflinks transcripts and RefSeq Chr19 reference, provided on the tutorial page. Was that the goal, or transcripts should overlap and something did not work? By the way, mapping for cufflinks was done to hg19, but reference annotation is provided at hg18. Maybe, this is the problem? Aleks Quoting Robert Curtis Hendrickson <curtish@uab.edu>:
Jen,
We are running into the same problem on our local install of galaxy. We're running Cufflinks v.1.0.1, on a BAM file (accepted_reads) from TopHat run on mm9 based RNAseq data (paired-end 25mer), and pulled down the changes made to galaxy last month to support the 1.0.1 version of Cufflinks.
We (think) we have mm9 indexes locally installed. We can successfully run get_genomic_sequence on mm9 .BED's....
Turning off bias correction made no difference.
We also tried rolling back to Cufflinks v0.9.1 (including the Galaxy patch), and got the same error
An error occurred running this job: cufflinks v0.9.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
An error occurred running this job: cufflinks v1.0.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
I can provide a link to the history on our server that you should (theoretically) be able to access.
Regards, Curtis
-----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user- bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: Friday, June 10, 2011 3:58 PM To: David Robinson Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] Error running cufflinks on Galaxy
Hello David,
Cufflinks requires locally cached data to perform the Bias Correction function.
Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem.
Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible).
Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine).
Jen Galaxy team
On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get an error:
An error occurred running this job: /cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from- uploaded-file
Thanks in advance for your help!
-David
************************************************
David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630
************************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
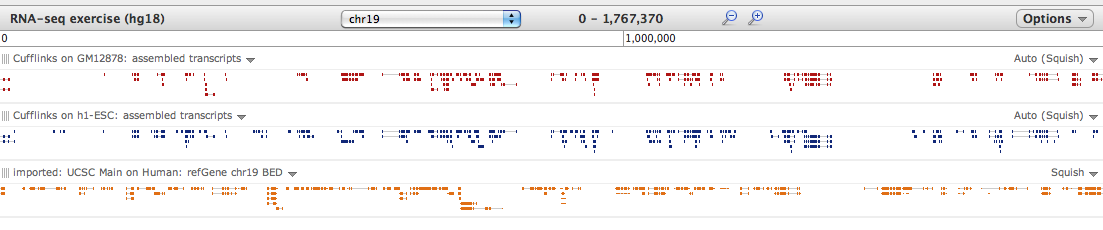
Hello Aleks, Thank you for bring these hg18/hg19 problem to our attention - it was introduced during an edit last week and has now been corrected. The good news is that results are obtained with either - the issue with overlap may be with the Visualization tool itself. When very "zoomed out", the overlap regions can vanish if small enough (we will be correcting/normalizing for this). However, the data is there if you just zoom in, as in the attached screenshot. Hopefully this helps! Best, Jen Galaxy team On 6/24/11 6:19 AM, Aleks Schein wrote:
Dear all,
I tried to follow the Cufflinks tutorial created by Jeremy. The problems start with first "visualization" step. The is no overlapping at all between Cufflinks transcripts and RefSeq Chr19 reference, provided on the tutorial page. Was that the goal, or transcripts should overlap and something did not work? By the way, mapping for cufflinks was done to hg19, but reference annotation is provided at hg18. Maybe, this is the problem?
Aleks
Quoting Robert Curtis Hendrickson <curtish@uab.edu>:
Jen,
We are running into the same problem on our local install of galaxy. We're running Cufflinks v.1.0.1, on a BAM file (accepted_reads) from TopHat run on mm9 based RNAseq data (paired-end 25mer), and pulled down the changes made to galaxy last month to support the 1.0.1 version of Cufflinks.
We (think) we have mm9 indexes locally installed. We can successfully run get_genomic_sequence on mm9 .BED's....
Turning off bias correction made no difference.
We also tried rolling back to Cufflinks v0.9.1 (including the Galaxy patch), and got the same error
An error occurred running this job: cufflinks v0.9.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
An error occurred running this job: cufflinks v1.0.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
I can provide a link to the history on our server that you should (theoretically) be able to access.
Regards, Curtis
-----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user- bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: Friday, June 10, 2011 3:58 PM To: David Robinson Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] Error running cufflinks on Galaxy
Hello David,
Cufflinks requires locally cached data to perform the Bias Correction function.
Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem.
Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible).
Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine).
Jen Galaxy team
On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get an error:
An error occurred running this job: /cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from- uploaded-file
Thanks in advance for your help!
-David
************************************************
David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630
************************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org/ http://galaxyproject.org/
{kind=link}
Thanks Jen, Another problem is with reference-guided assembly. When I produce Cufflinks assembled transcripts files and Cuffcompare combined transcrips file on my own data, it works fine with Galaxy. However, files cannot be visualized in UCSC browser-command "display at UCSC main" produces internal server error. When I try to upload them manually, I receive message "File xxx.gtf' - GFF/GTF group NM_005638 on chrX+, this line is on chrY+, all group members must be on same seq and strand " Any idea on what does it mean and how to fix it? Aleks Quoting Jennifer Jackson <jen@bx.psu.edu>:
Hello Aleks,
Thank you for bring these hg18/hg19 problem to our attention - it was introduced during an edit last week and has now been corrected.
The good news is that results are obtained with either - the issue with overlap may be with the Visualization tool itself. When very "zoomed out", the overlap regions can vanish if small enough (we will be correcting/normalizing for this). However, the data is there if you just zoom in, as in the attached screenshot.
Hopefully this helps!
Best,
Jen Galaxy team
On 6/24/11 6:19 AM, Aleks Schein wrote:
Dear all,
I tried to follow the Cufflinks tutorial created by Jeremy. The problems start with first "visualization" step. The is no overlapping at all between Cufflinks transcripts and RefSeq Chr19 reference, provided on the tutorial page. Was that the goal, or transcripts should overlap and something did not work? By the way, mapping for cufflinks was done to hg19, but reference annotation is provided at hg18. Maybe, this is the problem?
Aleks
Quoting Robert Curtis Hendrickson <curtish@uab.edu>:
Jen,
We are running into the same problem on our local install of galaxy. We're running Cufflinks v.1.0.1, on a BAM file (accepted_reads) from TopHat run on mm9 based RNAseq data (paired-end 25mer), and pulled down the changes made to galaxy last month to support the 1.0.1 version of Cufflinks.
We (think) we have mm9 indexes locally installed. We can successfully run get_genomic_sequence on mm9 .BED's....
Turning off bias correction made no difference.
We also tried rolling back to Cufflinks v0.9.1 (including the Galaxy patch), and got the same error
An error occurred running this job: cufflinks v0.9.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
An error occurred running this job: cufflinks v1.0.1 cufflinks -q --no-update-check -I 500000 -F 0.000100 -j 0.000100 -p 4 -N Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
I can provide a link to the history on our server that you should (theoretically) be able to access.
Regards, Curtis
-----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user- bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: Friday, June 10, 2011 3:58 PM To: David Robinson Cc: galaxy-user@lists.bx.psu.edu Subject: Re: [galaxy-user] Error running cufflinks on Galaxy
Hello David,
Cufflinks requires locally cached data to perform the Bias Correction function.
Without seeing any sample data, a quick guess is that changing the option "Tool: Cufflinks -> Perform Bias Correction:" from yes to no in that workflow step will probably correct the problem.
Another option is to set the dbkey value in the initial input FASTQ file to be a native database (if possible).
Hopefully this helps, but if does not correct the problem, please share a history link with data that demonstrates the problem and I can take closer look (emailing link to me directly, to maintain data privacy, would be fine).
Jen Galaxy team
On 6/8/11 12:07 PM, David Robinson wrote:
Hello,
When I attempt to run cufflinks based on .sam output from bowtie I get an error:
An error occurred running this job: /cufflinks v1.0.1 cufflinks -q --no-update-check -I 300000 -F 0.050000 -j 0.050000 -p 8 -b /galaxy/data/hg19/sam_index/hg19.fa Error running cufflinks. [Errno 2] No such file or directory: 'transcripts.gtf'
/What can I do to get around this problem and run cufflinks?
My workflow is on http://main.g2.bx.psu.edu and can be found here (I ran it using a .fastq file):
http://main.g2.bx.psu.edu/u/dgrtwo/w/cufflinks-workflow-imported-from- uploaded-file
Thanks in advance for your help!
-David
************************************************
David Robinson Graduate Student Lewis-Sigler Institute for Integrative Genomics Carl Icahn Laboratory Princeton University 646-620-6630
************************************************
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://usegalaxy.org/ http://galaxyproject.org/

Hi, I am working with HeLa cells and want to know how to get cufflinks etc. to highlight if a region of the genome is being transcribed that is not in the ensembl gtf. I know that cufflinks highlights with class code "j" regions that do not match a known gene and therefore may be novel but most of these arise from transcription on or near known genes. Does anyone know how to look for transcription that is clearly distinct from known genes? This is a wild goose chase but worth a peek just in case... Best Wishes, David. __________________________________ Dr David A. Matthews Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K. Tel. +44 117 3312058 Fax. +44 117 3312091 D.A.Matthews@bristol.ac.uk

Hi All, I was wondering if there is a way to add tools to the Galaxy web portal (the central server, not our own instance of Galaxy). Some of the tools I would like to use are described on "http://toolshed.g2.bx.psu.edu/repository?webapp=community". Can I add this functionality through my Galaxy account? Thanks Mete IMPORTANT WARNING: This email (and any attachments) is only intended for the use of the person or entity to which it is addressed, and may contain information that is privileged and confidential. You, the recipient, are obligated to maintain it in a safe, secure and confidential manner. Unauthorized redisclosure or failure to maintain confidentiality may subject you to federal and state penalties. If you are not the intended recipient, please immediately notify us by return email, and delete this message from your computer.

On Tue, Jul 5, 2011 at 11:41 PM, Mete Civelek <mcivelek@mednet.ucla.edu> wrote:
Hi All,
I was wondering if there is a way to add tools to the Galaxy web portal (the central server, not our own instance of Galaxy). Some of the tools I would like to use are described on "http://toolshed.g2.bx.psu.edu/repository?webapp=community". Can I add this functionality through my Galaxy account?
Thanks
Mete
No, you or your institute would have to setup a local Galaxy instance in order to add extra tools. This sort of thing is covered on the galaxy-dev mailing list. However, the Galaxy team might be willing to add popular tools to the main instance in some circumstances - so you can ask them. Peter
participants (8)
-
 admiral.david@gmail.com
admiral.david@gmail.com -
 Aleks Schein
Aleks Schein -
 David Matthews
David Matthews -
 David Robinson
David Robinson -
 Jennifer Jackson
Jennifer Jackson -
 Mete Civelek
Mete Civelek -
 Peter Cock
Peter Cock -
 Robert Curtis Hendrickson
Robert Curtis Hendrickson