What is the quality score type for the Solid datasets downloaded from SRA of NCBI?

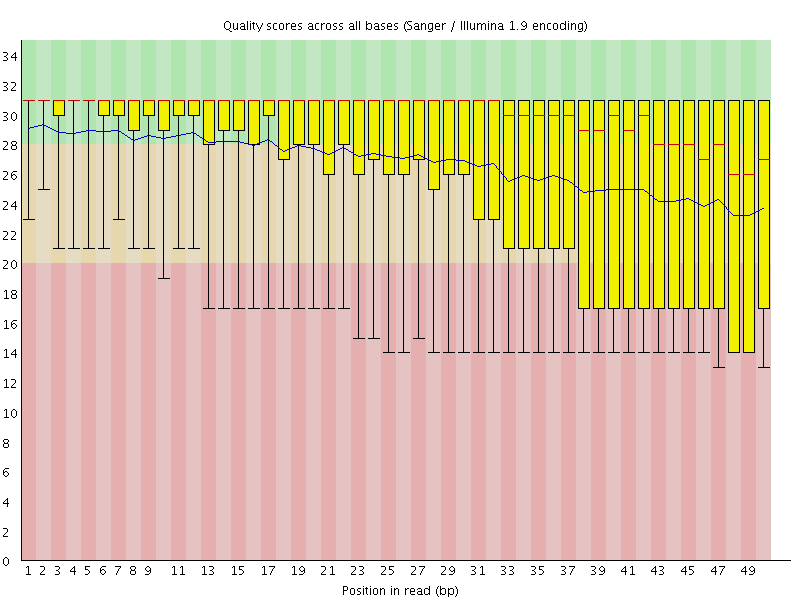
Hi all, Please help with the quality score type for the downloaded Solid datasets. I downloaded RNA-seq datasets, which were generated by AB Solid system, as base space and at FastQ format from SRA of NCBI. I uploaded the datasets onto the online sever Galaxy and change the datatype directly into "fastqsanger" and then test the quality by running FastQC. The output "per base quality" of solid dataset (please take look at the attached figure "per_base_quality-Solid") is quite different from the output "per base quality" of Illumina dataset (please compare with the attached figure "per base quality-Illumina"). The top score for Solid dataset is about 31, however the top score for Illumina dataset is 38. What is the quality score type for the downloaded Solid datasets when downloaded as base space and at FastQ format from SRA of NCBI? Please help me solve this problem. Thanks. Best regards. Jianguang Du
{kind=link}
{kind=link}

Hello Jianguang, The tool "NGS: QC and manipulation -> FASTQ Groomer" has some information about this, including a link to a wikipedia entry with more details specifically about the SRA: http://en.wikipedia.org/wiki/FASTQ_format http://en.wikipedia.org/wiki/FASTQ_format#NCBI_Sequence_Read_Archive And here is the SRA submission form, although the experimental record you downloaded from is the best place to find details: https://www.ebi.ac.uk/ena/about/sra_data_format SRA accepts CS and Fastq. In Galaxy these translate to: Color space reads: - datatype "Color Space Sanger" - annotated as "fastqcssanger" Fastq reads: - datatype with Phred quality offset 64 "Illumina 1.3-1.7" - annotated as "fastqillumina" and - datatype with Phred quality offset 33 "Illumina 1.8+" - annotated as "fastqsanger" Many tools require "fastqsanger". Use the "FASTQ Groomer" to transform as needed, but double check with FastQC just like you are doing. I have seen data labeled as Illumina 1.5 that was really already scaled to Phred+33, or at least appeared to be. In the end this is a judgement call or you can try to contact SRA/data authors for a definitive answer if there are no processing notes in the experiment (often the case). Hopefully this helps, Jen Galaxy team On 3/5/13 8:18 AM, Gene Genome wrote:
Hi all,
Please help with the quality score type for the downloaded Solid datasets. I downloaded RNA-seq datasets, which were generated by AB Solid system, as base space and at FastQ format from SRA of NCBI. I uploaded the datasets onto the online sever Galaxy and change the datatype directly into "fastqsanger" and then test the quality by running FastQC. The output "per base quality" of solid dataset (please take look at the attached figure "per_base_quality-Solid") is quite different from the output "per base quality" of Illumina dataset (please compare with the attached figure "per base quality-Illumina"). The top score for Solid dataset is about 31, however the top score for Illumina dataset is 38. What is the quality score type for the downloaded Solid datasets when downloaded as base space and at FastQ format from SRA of NCBI? Please help me solve this problem. Thanks. Best regards. Jianguang Du
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org
participants (2)
-
 Gene Genome
Gene Genome -
 Jennifer Jackson
Jennifer Jackson