Re: [galaxy-user] tophat to cuffdiff without cufflinks

Well in that case I will like to use the same gtf file from TAIR 10 that was used by the tophat. But the tophat have the option bult in. How can I point cuffdiff to the same file? Dr. Alejandro Colaneri Departments of Biology University of North Carolina at Chapel Hill 310 Coker Hall, CB# 3280 120 South Road Chapel Hill, NC 27599-3280 Tel: 919-962-2273 fax: 919- 962-1625 From: Jennifer Jackson <jen@bx.psu.edu<mailto:jen@bx.psu.edu>> Date: Thursday, May 30, 2013 8:07 PM To: "Colaneri, Alejandro Cesar" <colaneri@email.unc.edu<mailto:colaneri@email.unc.edu>> Cc: "galaxy-user@bx.psu.edu<mailto:galaxy-user@bx.psu.edu>" <galaxy-user@bx.psu.edu<mailto:galaxy-user@bx.psu.edu>> Subject: Re: [galaxy-user] tophat to cuffdiff without cufflinks Hello, You will need to provide a GTF or GFF3 file to Cuffdiff - this is what the tool uses as a reference base to build gene, transcript, and if provided in the annotation attributes, transcript start site and protein groupings to perform the differential analysis. More details can be found here: http://cufflinks.cbcb.umd.edu/manual.html Our tutorial and other wiki help is linked from here, see "Tools on the Main Server": http://wiki.galaxyproject.org/Support#Interpreting_scientific_results Hopefully this helps, Jen Galaxy team On 5/30/13 3:15 PM, Colaneri, Alejandro Cesar wrote: Hi I comparing gen expression data (RNA-seq) in Arabidopsis. Different genotypes, different conditions. Since Arabidopsis is very well annotated I decided to do cuffdiff directly after tophat. However when building my workflow I found that the cuffdiff said a gtf file is necessary. Please see the picture in this email, under INPUT FORMAT. My question is if I can still compare my libraries in the way I designed below. [cid:part1.04070709.03020802@bx.psu.edu] Dr. Alejandro Colaneri Departments of Biology University of North Carolina at Chapel Hill 310 Coker Hall, CB# 3280 120 South Road Chapel Hill, NC 27599-3280 Tel: 919-962-2273 fax: 919- 962-1625 ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/ -- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org
{kind=link}

Hello, Perhaps there is a mix-up about a reference annotation (GTF/GFF3) resource versus a reference genome database? By "built-in", do you mean the TAIR 10 reference genome that Tophat can use as a target genomic sequence database? If so, then that can also be specified when running Cuffdiff (this is probably a good idea to do), but a reference annotation resource (genes and transcripts, not genomic sequence) is still needed to perform the differential expression. I haven't seen your history, so if you did use a reference annotation GTF/GFF3 file with Tophat (this is possible, as an additional input, when using "Advanced parameters" - along with the reference genome's genomic database), then you can and should definitely use that same one again. But, it wouldn't be "built-in", instead would be a dataset uploaded by you, and already in your history - so I am making a guess that this is not what was used. But I may have misunderstood. When you do locate a reference annotation dataset (if you do not already have one), be sure to review the RNA-seq FAQ linked from the wiki I sent earlier, as you may need to adjust the chromosome identifiers in the file before using it - they have to be formatted exactly like the genomic chromosome identifiers to work correctly with the tool. You can run a tool like Picard's " BAM Index Statistics" on the Tophat output to get the chromosome formats from the built-in Arabidopsis reference genome, compare that to the reference annotation file you have, and make adjustments as needed following the general guidelines in the FAQ, using tools in the group "Text Manipulation". Since the genome is not mammalian (the chrom formats are slightly different), the processing will likely not be identical to that in the FAQ, but the overall workflow will be similar. This is easy to mix up, so hopefully some part of this helps to clarify the different inputs, Jen Galaxy team On 5/30/13 7:50 PM, Colaneri, Alejandro Cesar wrote:
Well in that case I will like to use the same gtf file from TAIR 10 that was used by the tophat. But the tophat have the option bult in. How can I point cuffdiff to the same file?
//
//
//
/Dr. Alejandro Colaneri/
/
Departments of Biology
University of North Carolina at Chapel Hill
310 Coker Hall, CB# 3280
120 South Road
Chapel Hill, NC 27599-3280
Tel: 919-962-2273
fax: 919- 962-1625
/ //
From: Jennifer Jackson <jen@bx.psu.edu <mailto:jen@bx.psu.edu>> Date: Thursday, May 30, 2013 8:07 PM To: "Colaneri, Alejandro Cesar" <colaneri@email.unc.edu <mailto:colaneri@email.unc.edu>> Cc: "galaxy-user@bx.psu.edu <mailto:galaxy-user@bx.psu.edu>" <galaxy-user@bx.psu.edu <mailto:galaxy-user@bx.psu.edu>> Subject: Re: [galaxy-user] tophat to cuffdiff without cufflinks
Hello,
You will need to provide a GTF or GFF3 file to Cuffdiff - this is what the tool uses as a reference base to build gene, transcript, and if provided in the annotation attributes, transcript start site and protein groupings to perform the differential analysis.
More details can be found here: http://cufflinks.cbcb.umd.edu/manual.html
Our tutorial and other wiki help is linked from here, see "Tools on the Main Server": http://wiki.galaxyproject.org/Support#Interpreting_scientific_results
Hopefully this helps,
Jen Galaxy team
On 5/30/13 3:15 PM, Colaneri, Alejandro Cesar wrote:
Hi
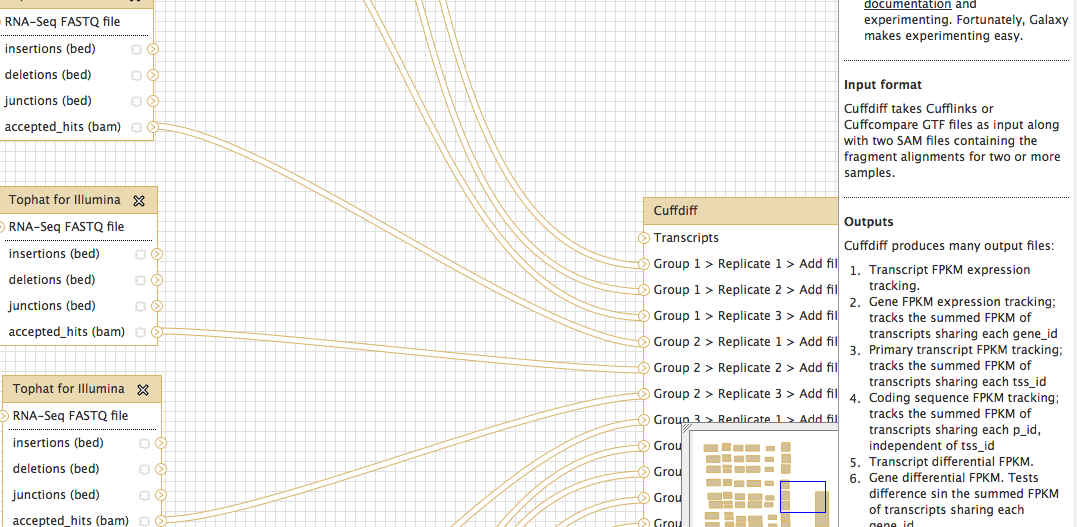
I comparing gen expression data (RNA-seq) in Arabidopsis. Different genotypes, different conditions. Since Arabidopsis is very well annotated I decided to do cuffdiff directly after tophat. However when building my workflow I found that the cuffdiff said a gtf file is necessary. Please see the picture in this email, under INPUT FORMAT. My question is if I can still compare my libraries in the way I designed below.
//
//
//
/Dr. Alejandro Colaneri/
/
Departments of Biology
University of North Carolina at Chapel Hill
310 Coker Hall, CB# 3280
120 South Road
Chapel Hill, NC 27599-3280
Tel: 919-962-2273
fax: 919- 962-1625
///
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
-- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org
-- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org
participants (2)
-
 Colaneri, Alejandro Cesar
Colaneri, Alejandro Cesar -
 Jennifer Jackson
Jennifer Jackson