
Dear galaxy users, We have done deep sequencing on some known genomic loci using Hiseq2000. I have already mapped the reads to the reference sequences by using Galaxy. In the next step, I want to find SNPs and calculate the SNP percentage within the reads. There are 500,000 to 1,000,000 reads per biological sample. Can I do it with galaxy? If not, is there other programs available in windows? Considering that I am not very familiar with programming. Thanks, Xiefan University of Florida
Dear galaxy users, We have done deep sequencing on some known genomic loci using Hiseq2000. I have already mapped the reads to the reference sequences by using Galaxy. In the next step, I want to find SNPs and calculate the SNP percentage within the reads. There are 500,000 to 1,000,000 reads per biological sample. Can I do it with galaxy? If not, is there other programs available in windows? Considering that I am not very familiar with programming. Thanks, Xiefan University of Florida

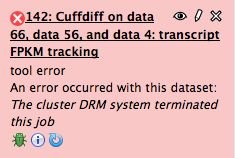
Dear Galaxy Users I wondered if anybody is having the same problem. I am trying to run CuffDiff using the latest version of GenCode18 and keep getting the following error message. Is there a problem with the network or is it me? Best wishes Mark
{kind=link}

Hi Mark, This error is not tool related. Could you click the “bug” icon to send us an error report? —nate On Nov 11, 2013, at 8:33 AM, Mark Lindsay <m.a.lindsay@bath.ac.uk> wrote:
Dear Galaxy Users
I wondered if anybody is having the same problem.
I am trying to run CuffDiff using the latest version of GenCode18 and keep getting the following error message.
Is there a problem with the network or is it me?
Best wishes
Mark
<Screen Shot 2013-11-11 at 13.32.12.png> ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:

Hello Xiefan, This page has a simple breakdown of how to call variants within Galaxy. This is brand new, so full annotation/video is pending, but it should still be straightforward to see how the data is prepped and which tools are used. https://usegalaxy.org/u/galaxyproject/p/galaxy-101-ngs-variant Another set of tutorial videos cover the tools, which you can use on the public site or on a scaled up cloud server as needed: https://vimeo.com/channels/galaxytoolshed Hopefully this help you to get started. Jen Galaxy team On 10/21/12 3:11 PM, Xiefan Fang wrote:
Dear galaxy users, We have done deep sequencing on some known genomic loci using Hiseq2000. I have already mapped the reads to the reference sequences by using Galaxy. In the next step, I want to find SNPs and calculate the SNP percentage within the reads. There are 500,000 to 1,000,000 reads per biological sample. Can I do it with galaxy? If not, is there other programs available in windows? Considering that I am not very familiar with programming.
Thanks, Xiefan University of Florida
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Hillman-Jackson http://galaxyproject.org

Hi all, First of all, thank you again for all your efforts to develop a project like Galaxy, that makes leading-edge bioinformatic tools available for non-bioinformaticians and wet-lab biologists like me ! I am using GATK Unified Geneotyper through the Galaxy main server to analyze variations from whole-genome re-sequencing data. I have read in the GATK documentation that there is a tool called "CallableLoci", that gives a .bed file of the genome indicating specifically which base where callable or not by Unified Genotyper (UG), using different criteria such as read depth, base quality, mapping quality. The log & metrics files generated by UG in Galaxy give the general statistics of callable loci, but there is no such a file giving a detailed information of the eligibility of each base. Right now I am using the tool "depth of coverage on BAM file" to get an idea of this information, but it's only partial since it doesn't take into account all the parameters used by UG to consider a locus callable (notably base quality and mapping quality). Do you think it would be possible to implement the "CallableLoci" tool in Galaxy? Would someone propose me an alternative to get this precious information on which areas of the genome are considered callable ? Thanks for your help/advice, Fabrice
Hi all, Would someone know how to get the information on which areas of the genome are considered callable after a call with Unified genotyper from GATK (as a .bed or pileup file)? Thanks for your help/advice, Fabrice -- Fabrice Besnard Institute of Biology of the Ecole Normale Supérieure (IBENS) 46 rue d'Ulm, 75230 Paris cedex 05, France 8th floor. Office: Room 802. Lab: Room 817. mail: fbesnard@biologie.ens.fr Tel: +33-1-44-32-39-31
Hi Fabrice, I know I already replied to your other post about this tool and the status/reasons why it is not wrapped in Galaxy right now, but I was going to suggest that you ask on the GATK forum for help about using the "CallableLoci" tool on the command-line since you asked again. Then I checked for prior Q/A and I saw that you were a step ahead and already found advice on 11/8: http://gatkforums.broadinstitute.org/discussion/3448/unified-genotyper-with-... This really is the best advice, including the coverage part of the question there. As long as you are an acedemic/non-profit user, there should be no licensing issues using the tool directly. I didn't see anything posted at seqanswers, and that can often be another source of information, but I would have to agree with Geraldine that going command-line with the "CallableLoci" tool is probably your best option since this is the exact data that you want, and I am not aware of any web-based implementations. But, this is still open for others to comment on! The web is a big place & all input is welcome. Good luck with your project! Jen Galaxy team On 11/25/13 8:59 AM, Fabrice Besnard wrote:
Hi all,
Would someone know how to get the information on which areas of the genome are considered callable after a call with Unified genotyper from GATK (as a .bed or pileup file)?
Thanks for your help/advice,
Fabrice
-- Jennifer Hillman-Jackson http://galaxyproject.org
Hi Jen, Thanks a lot for spending time again on my issue ! I think I will go for command-line solution ! Thanks, Fabrice Le 03/12/2013 00:43, Jennifer Jackson a écrit :
Hi Fabrice,
I know I already replied to your other post about this tool and the status/reasons why it is not wrapped in Galaxy right now, but I was going to suggest that you ask on the GATK forum for help about using the "CallableLoci" tool on the command-line since you asked again. Then I checked for prior Q/A and I saw that you were a step ahead and already found advice on 11/8: http://gatkforums.broadinstitute.org/discussion/3448/unified-genotyper-with-...
This really is the best advice, including the coverage part of the question there. As long as you are an acedemic/non-profit user, there should be no licensing issues using the tool directly.
I didn't see anything posted at seqanswers, and that can often be another source of information, but I would have to agree with Geraldine that going command-line with the "CallableLoci" tool is probably your best option since this is the exact data that you want, and I am not aware of any web-based implementations.
But, this is still open for others to comment on! The web is a big place & all input is welcome.
Good luck with your project!
Jen Galaxy team
On 11/25/13 8:59 AM, Fabrice Besnard wrote:
Hi all,
Would someone know how to get the information on which areas of the genome are considered callable after a call with Unified genotyper from GATK (as a .bed or pileup file)?
Thanks for your help/advice,
Fabrice
-- Fabrice Besnard Institute of Biology of the Ecole Normale Supérieure (IBENS) 46 rue d'Ulm, 75230 Paris cedex 05, France 8th floor. Office: Room 802. Lab: Room 817. mail: fbesnard@biologie.ens.fr Tel: +33-1-44-32-39-31
Hi Fabrice, This tool is not implemented in Galaxy as it was released after the current GATK (beta) tool group was created. There is some development on the last GATK-lite pipeline, but as you know this does not include the latest version of all tools. The public Main Galaxy server (usegalaxy.org) only contains tools that are licensed for general use, no restrictions. One option is to use the tool command-line, then upload the results into Galaxy for use with our other tools to compliment/incorporate it into your larger analysis. There is much help offered on the GATK forum, and this is just one tool. So, even if you are experienced with bioinformatics on the command-line, this might be possible. You seem to be very knowledgeable about these tools and how they function! My guess is that you'll be able to pick this up! Sorry that we could not help more directly right now. But please follow this Trello ticket to watch upcoming GATK tool wrapper development by the Galaxy team. If this specific tool does turn out to be added in the lite implementation, this is a great way to know when to look for it: https://trello.com/c/IPkT2spd Best! Jen Galaxy team On 11/20/13 2:25 AM, Fabrice Besnard wrote:
Hi all,
First of all, thank you again for all your efforts to develop a project like Galaxy, that makes leading-edge bioinformatic tools available for non-bioinformaticians and wet-lab biologists like me !
I am using GATK Unified Geneotyper through the Galaxy main server to analyze variations from whole-genome re-sequencing data. I have read in the GATK documentation that there is a tool called "CallableLoci", that gives a .bed file of the genome indicating specifically which base where callable or not by Unified Genotyper (UG), using different criteria such as read depth, base quality, mapping quality. The log & metrics files generated by UG in Galaxy give the general statistics of callable loci, but there is no such a file giving a detailed information of the eligibility of each base.
Right now I am using the tool "depth of coverage on BAM file" to get an idea of this information, but it's only partial since it doesn't take into account all the parameters used by UG to consider a locus callable (notably base quality and mapping quality).
Do you think it would be possible to implement the "CallableLoci" tool in Galaxy? Would someone propose me an alternative to get this precious information on which areas of the genome are considered callable ?
Thanks for your help/advice,
Fabrice
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
-- Jennifer Hillman-Jackson http://galaxyproject.org
participants (5)
-
 Fabrice Besnard
Fabrice Besnard -
 Jennifer Jackson
Jennifer Jackson -
 Mark Lindsay
Mark Lindsay -
 Nate Coraor
Nate Coraor -
 Xiefan Fang
Xiefan Fang