Why don't we get FPKMs from this gene?

Hello, We are using BAM to map Illumina reads to a bacterial genome, followed by Cufflinks o get the FPKMs. We have come across many genes for which we get FPKM=0 (using both gene and transcript expression) even though there are reads mapping to these gene IDs (e.g. the region between the dashed lines in the attached screenshot). Can anyone suggest a reason/fix for this? Thanks Dikla

{kind=link}

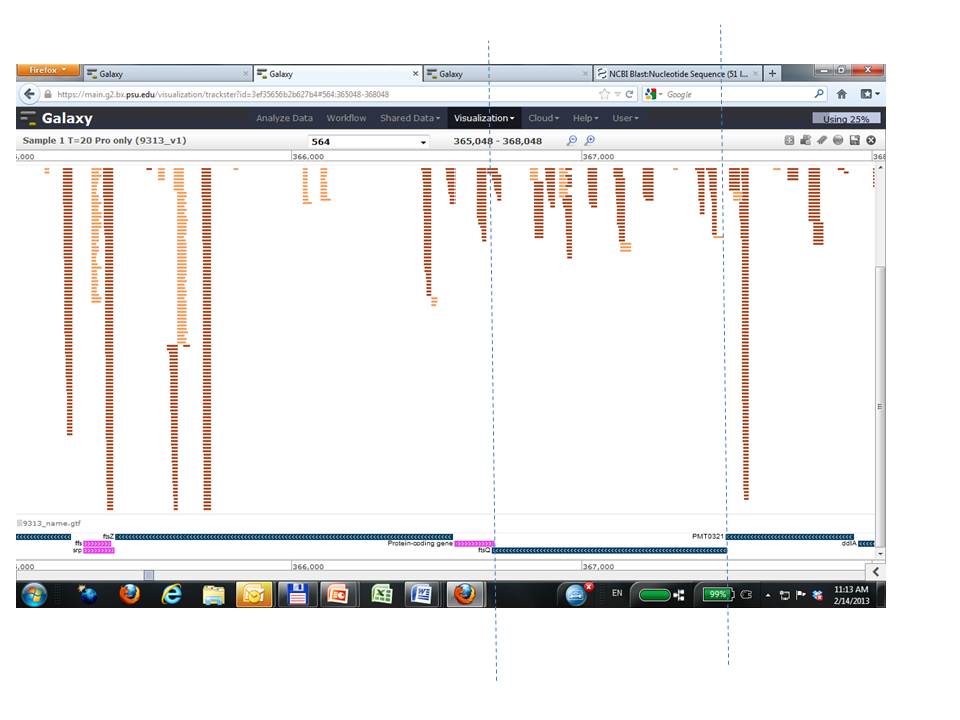
Hello Dikla, Information in the sixth field of these same files will provide information about why the calculations for individual genes/transcripts were not performed. Possible values are explained in the tool documentation: http://cufflinks.cbcb.umd.edu/manual.html#gene_exp_diff This particular region appears to have low coverage compared to the surrounding regions (e.g. low abundance), but this is of course only a small sample, and it is difficult to know about other criteria considered by the tool from the graphic (proper pairing, multiple map locations, etc.). But if you believe that higher abundance transcripts are preventing lower abundance transcripts from being evaluated, or even just suspect that and want to test, you could try running Cufflinks with the option "Perform quartile normalization: Yes". Using "Perform Bias Correction: Yes" is also another parameter to explore (requires a reference genome). The Cufflinks web site is a great resource to learn more about these parameters and the Galaxy tool form has each included as an option. Hopefully this helps, Jen Galaxy team On 2/14/13 1:48 AM, Dikla Aharonovich wrote:
Hello,
We are using BAM to map Illumina reads to a bacterial genome, followed by Cufflinks o get the FPKMs. We have come across many genes for which we get FPKM=0 (using both gene and transcript expression) even though there are reads mapping to these gene IDs (e.g. the region between the dashed lines in the attached screenshot). Can anyone suggest a reason/fix for this?
Thanks
Dikla
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org

Hello Jen, Thanks for your reply and input. The genes which have"0 FPKM" have different tags: some are LOWDATA, some are HIDATA and some are OK... We tried changing the number of reads used as input into Cufflinks - when we used many reads (~12,000,000 on a genome with ~2,400 genes) most of the genes have 0 FPKM and "HIDATA". When we used fewer reads (1,000,000) on the same genome many of the genes still had "0 FPKM" but now with different flags (OK or LOWDATA). We tried using quartile normalization and this didn't seem to help much. As in the previous cases, looking at the reads using IGV showed that genes with 0 FPKM, even if they have "OK" as their tag, do indeed have reads associated with them. Any suggestions? Could this be due to the fact that these are bacterial genomes without introns? If so, any suggestions what parameters to change? Thanks Dikla and Daniel -----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user-bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: שבת 23 פברואר 2013 11:03 To: דקלה אהרונוביץ Cc: דניאל שר; galaxy-user@bx.psu.edu Subject: Re: [galaxy-user] Why don't we get FPKMs from this gene? Hello Dikla, Information in the sixth field of these same files will provide information about why the calculations for individual genes/transcripts were not performed. Possible values are explained in the tool documentation: http://cufflinks.cbcb.umd.edu/manual.html#gene_exp_diff This particular region appears to have low coverage compared to the surrounding regions (e.g. low abundance), but this is of course only a small sample, and it is difficult to know about other criteria considered by the tool from the graphic (proper pairing, multiple map locations, etc.). But if you believe that higher abundance transcripts are preventing lower abundance transcripts from being evaluated, or even just suspect that and want to test, you could try running Cufflinks with the option "Perform quartile normalization: Yes". Using "Perform Bias Correction: Yes" is also another parameter to explore (requires a reference genome). The Cufflinks web site is a great resource to learn more about these parameters and the Galaxy tool form has each included as an option. Hopefully this helps, Jen Galaxy team On 2/14/13 1:48 AM, Dikla Aharonovich wrote:
Hello,
We are using BAM to map Illumina reads to a bacterial genome, followed by Cufflinks o get the FPKMs. We have come across many genes for which we get FPKM=0 (using both gene and transcript expression) even though there are reads mapping to these gene IDs (e.g. the region between the dashed lines in the attached screenshot). Can anyone suggest a reason/fix for this?
Thanks
Dikla
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
Hi again Jen, We got the following reply to this question (why do we get FPKM=0 and HIDATA with Cufflinks) on SeqAnswers: "From the Cufflinks manual: --max-bundle-frags <int> Sets the maximum number of fragments a locus may have before being skipped. Skipped loci are marked with status HIDATA. Default: 1000000 Just make that option higher. You will need to be sure the high number of reads mapping there represent reality rather than some sort of artifact though." What value does Galaxy use for -max-bundle-frags? We could not find a way of changing this parameter. Thanks Dikla and Daniel -----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user-bounces@lists.bx.psu.edu] On Behalf Of Daniel Sher Sent: יום ב 04 מרץ 2013 14:17 To: 'Jennifer Jackson'; דקלה אהרונוביץ Cc: galaxy-user@bx.psu.edu Subject: Re: [galaxy-user] Why don't we get FPKMs from this gene? Hello Jen, Thanks for your reply and input. The genes which have"0 FPKM" have different tags: some are LOWDATA, some are HIDATA and some are OK... We tried changing the number of reads used as input into Cufflinks - when we used many reads (~12,000,000 on a genome with ~2,400 genes) most of the genes have 0 FPKM and "HIDATA". When we used fewer reads (1,000,000) on the same genome many of the genes still had "0 FPKM" but now with different flags (OK or LOWDATA). We tried using quartile normalization and this didn't seem to help much. As in the previous cases, looking at the reads using IGV showed that genes with 0 FPKM, even if they have "OK" as their tag, do indeed have reads associated with them. Any suggestions? Could this be due to the fact that these are bacterial genomes without introns? If so, any suggestions what parameters to change? Thanks Dikla and Daniel -----Original Message----- From: galaxy-user-bounces@lists.bx.psu.edu [mailto:galaxy-user-bounces@lists.bx.psu.edu] On Behalf Of Jennifer Jackson Sent: שבת 23 פברואר 2013 11:03 To: דקלה אהרונוביץ Cc: דניאל שר; galaxy-user@bx.psu.edu Subject: Re: [galaxy-user] Why don't we get FPKMs from this gene? Hello Dikla, Information in the sixth field of these same files will provide information about why the calculations for individual genes/transcripts were not performed. Possible values are explained in the tool documentation: http://cufflinks.cbcb.umd.edu/manual.html#gene_exp_diff This particular region appears to have low coverage compared to the surrounding regions (e.g. low abundance), but this is of course only a small sample, and it is difficult to know about other criteria considered by the tool from the graphic (proper pairing, multiple map locations, etc.). But if you believe that higher abundance transcripts are preventing lower abundance transcripts from being evaluated, or even just suspect that and want to test, you could try running Cufflinks with the option "Perform quartile normalization: Yes". Using "Perform Bias Correction: Yes" is also another parameter to explore (requires a reference genome). The Cufflinks web site is a great resource to learn more about these parameters and the Galaxy tool form has each included as an option. Hopefully this helps, Jen Galaxy team On 2/14/13 1:48 AM, Dikla Aharonovich wrote:
Hello,
We are using BAM to map Illumina reads to a bacterial genome, followed by Cufflinks o get the FPKMs. We have come across many genes for which we get FPKM=0 (using both gene and transcript expression) even though there are reads mapping to these gene IDs (e.g. the region between the dashed lines in the attached screenshot). Can anyone suggest a reason/fix for this?
Thanks
Dikla
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
participants (4)
-
 Daniel Sher
Daniel Sher -
 Dikla Aharonovich
Dikla Aharonovich -
 Jennifer Jackson
Jennifer Jackson -
 Noa Sher
Noa Sher