Count reads mapping to introns, extragenic regions

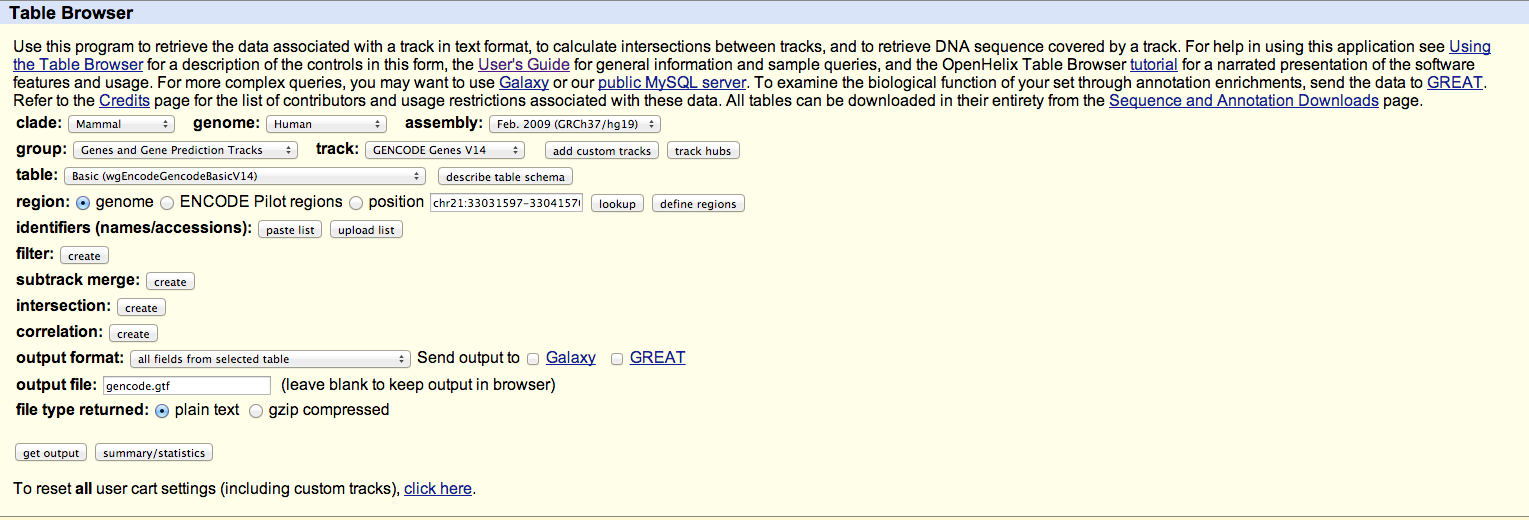
Hello everyone, I have some SAM/BAM files containing the alignments of some RNA-seq reads to hg19. I'm interested in calculating some mapping statistics, specifically, the percentage of reads mapping to exons, introns, and extragenic regions. I gather that this can be done with bedtools, but I'm finding myself a little bit stuck just figuring out what files I need to get this information. I gather that I need a GTF (or possibly GFF) file, and I downloaded one from the UCSC browser using the settings in the attached image. The first couple lines of the resulting file are pasted below. I see that the file has exon start and end sites. Is there a way to get what I need with this file, or do I need something else? Any assistance would be much appreciated, Thanks Alex cat gencode.gtf | head -3 #bin name chrom strand txStart txEnd cdsStart cdsEnd exonCount exonStarts exonEnds score name2 cdsStartStat cdsEndStat exonFrames 0 ENST00000237247.6 chr1 + 66999065 67210057 67000041 67208778 27 66999065,66999928,67091529,67098752,67099762,67105459,67108492,67109226,67126195,67133212,67136677,67137626,67138963,67142686,67145360,67147551,67149789,67154830,67155872,67161116,67184976,67194946,67199430,67205017,67206340,67206954,67208755, 66999090,67000051,67091593,67098777,67099846,67105516,67108547,67109402,67126207,67133224,67136702,67137678,67139049,67142779,67145435,67148052,67149870,67154958,67155999,67161176,67185088,67195102,67199563,67205220,67206405,67207119,67210057, 0 SGIP1 cmpl cmpl -1,0,1,2,0,0,0,1,0,0,0,1,2,1,1,1,1,1,0,1,1,2,2,0,2,1,1, 0 ENST00000371039.1 chr1 + 66999274 67210768 67000041 67208778 22 66999274,66999928,67091529,67098752,67105459,67108492,67109226,67136677,67137626,67138963,67142686,67145360,67154830,67155872,67160121,67184976,67194946,67199430,67205017,67206340,67206954,67208755, 66999355,67000051,67091593,67098777,67105516,67108547,67109402,67136702,67137678,67139049,67142779,67145435,67154958,67155999,67160187,67185088,67195102,67199563,67205220,67206405,67207119,67210768, 0 SGIP1 cmpl cmpl -1,0,1,2,0,0,1,0,1,2,1,1,1,0,1,1,2,2,0,2,1,1,
{kind=link}

Hi Alex, It may be simplest to extract the comparison coordinates from the UCSC Table browser in the groups you wish to count statistics on directly, in BED format, rather than in GTF format and parsing from there. You can also send the data directly to your history by checking the "Galaxy" box next in the send output section. This avoids having to download/upload the files. I don't think that BEDTools are going to help when teasing apart UTR, intron, and exon regions when using a BED12. There is the tool " Filter and Sort -> Extract features from GFF data" that will parse a BED12 file, but again, the Table Browser will do this for you directly, so I would recommended that. Then, once you have the different group's coordinates in BED6 format in Galaxy, the tools in "Operate on Genomic Intervals" will probably be the most helpful. Extract the coordinates from SAM/BAM files (if needed) with "NGS: SAM Tools -> Convert SAM to interval". This is the general path to get the comparison regions from the Table Browser: Start in Galaxy from the working history. Click through "Get Data -> UCSC Main" link, at the top of the left tool panel, to reach the Table Browser. Set up the extract from a track in the "Gene and Gene Predictions" group and choose BED for the output. Galaxy will be checked by default as an output. Do not name an output file, leave that blank and submit. On the next form (you will still be in the Table Browser), the lower section will have a list of genomic regions. You can check the boxes to mix regions into clusters that fit the groups you want to gather statistics for. Keep in mind that you can choose regions to be used in "negative" comparisons here. E.g. when comparing two coordinate sets it is possible to select those with "no overlap". Be sure to see the help on the Interval tools - each explains the operation in detail. Best, Jen Galaxy team On 3/1/13 6:31 AM, Alex Koeppel wrote:
Hello everyone,
I have some SAM/BAM files containing the alignments of some RNA-seq reads to hg19. I'm interested in calculating some mapping statistics, specifically, the percentage of reads mapping to exons, introns, and extragenic regions. I gather that this can be done with bedtools, but I'm finding myself a little bit stuck just figuring out what files I need to get this information. I gather that I need a GTF (or possibly GFF) file, and I downloaded one from the UCSC browser using the settings in the attached image.
The first couple lines of the resulting file are pasted below. I see that the file has exon start and end sites. Is there a way to get what I need with this file, or do I need something else?
Any assistance would be much appreciated,
Thanks
Alex
cat gencode.gtf | head -3 #bin name chrom strand txStart txEnd cdsStart cdsEnd exonCount exonStarts exonEnds score name2 cdsStartStat cdsEndStat exonFrames 0 ENST00000237247.6 chr1 + 66999065 67210057 67000041 67208778 27 66999065,66999928,67091529,67098752,67099762,67105459,67108492,67109226,67126195,67133212,67136677,67137626,67138963,67142686,67145360,67147551,67149789,67154830,67155872,67161116,67184976,67194946,67199430,67205017,67206340,67206954,67208755, 66999090,67000051,67091593,67098777,67099846,67105516,67108547,67109402,67126207,67133224,67136702,67137678,67139049,67142779,67145435,67148052,67149870,67154958,67155999,67161176,67185088,67195102,67199563,67205220,67206405,67207119,67210057, 0 SGIP1 cmpl cmpl -1,0,1,2,0,0,0,1,0,0,0,1,2,1,1,1,1,1,0,1,1,2,2,0,2,1,1, 0 ENST00000371039.1 chr1 + 66999274 67210768 67000041 67208778 22 66999274,66999928,67091529,67098752,67105459,67108492,67109226,67136677,67137626,67138963,67142686,67145360,67154830,67155872,67160121,67184976,67194946,67199430,67205017,67206340,67206954,67208755, 66999355,67000051,67091593,67098777,67105516,67108547,67109402,67136702,67137678,67139049,67142779,67145435,67154958,67155999,67160187,67185088,67195102,67199563,67205220,67206405,67207119,67210768, 0 SGIP1 cmpl cmpl -1,0,1,2,0,0,1,0,1,2,1,1,1,0,1,1,2,2,0,2,1,1,
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Hillman-Jackson Galaxy Support and Training http://galaxyproject.org
participants (2)
-
 Alex Koeppel
Alex Koeppel -
 Jennifer Jackson
Jennifer Jackson