Re: [galaxy-user] Genome Browser Histogram Visualization of Accepted Hits

Hi Trent, I realized that part of your question went unanswered: On 5/31/12 1:58 PM, Fowler, Trent wrote:
Dear Jen,
Well, it took ~4 hours to load but the visualization track is working and I am able to show 5 tracks on the screen and scan different genes. All quite good. However, I cannot find how to copy/paste or save the graphs for presentation in a figure. Afterwards, such a figure could be lined up with the gene structure a la UCSC GB. On that note, your note mentioned something…
If there is a particular UCSC track (or another source mapped to same reference genome) that you wish to compare your data against, it could be imported into Galaxy and added to your Trackster visualization session.
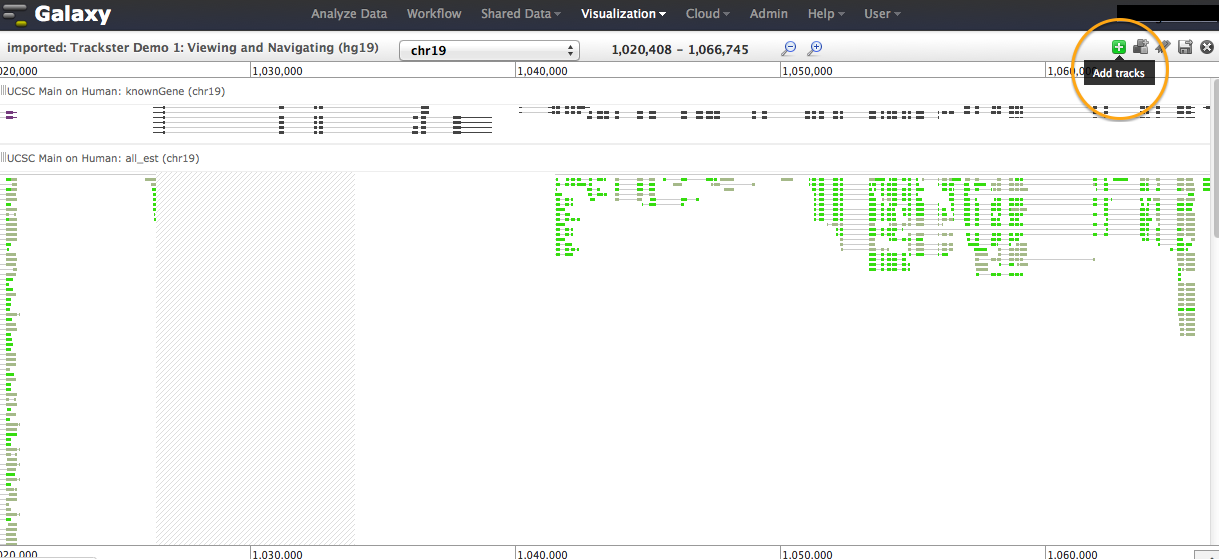
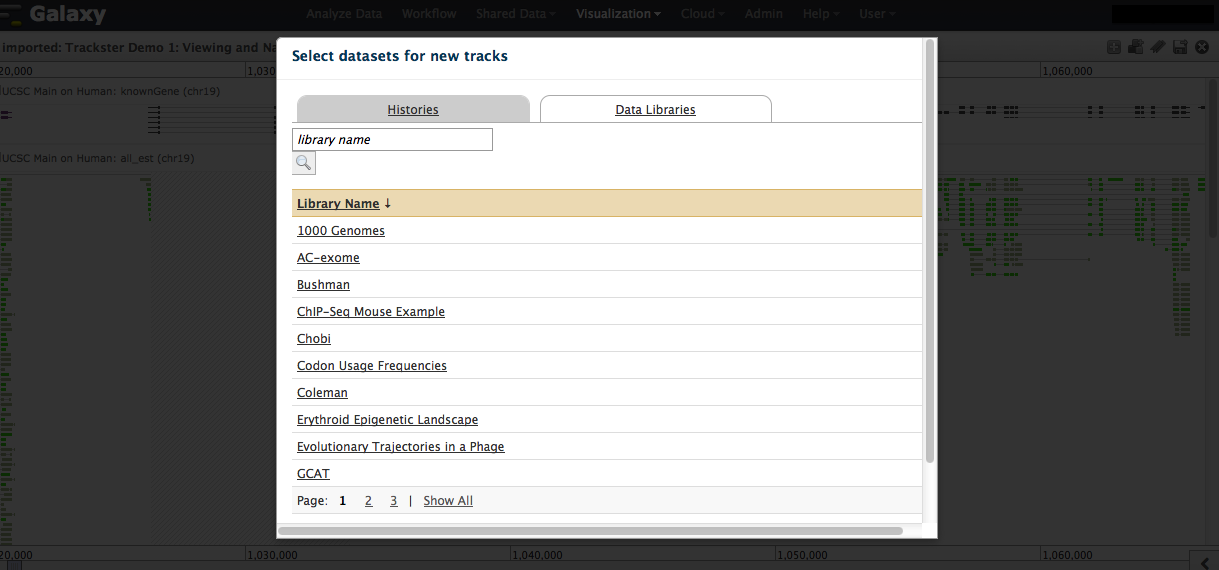
Any track loaded into any of your histories can be added to a visualization. Click on the "plus" icon to add tracks. See addtrack1.png. You will be able to navigate by default through your active history, but can also move to any of your histories or the Shared Data Libraries, to select datasets (one or more) and import. See addtrack2.png. The visualization tool recently went through an intensive development cycle. Documentation/tutorials to cover the full functionality is coming. Until then, please let us know if you need pointers when exploring the UI. Best, Jen Galaxy team
Is there a reference somewhere on how to do this?
Best,
Trent
*From:*Jennifer Jackson [mailto:jen@bx.psu.edu] *Sent:* Wednesday, May 30, 2012 3:17 PM *To:* Fowler, Trent *Cc:* galaxy-user@bx.psu.edu *Subject:* Re: [galaxy-user] Genome Browser Histogram Visualization of Accepted Hits
Hi Trent,
Hiram's advice is very good for UCSC display (thanks Hiram!)
You most likely heard of converting BAM->SAM->Interval->BED->BigBED. But, Hiram is exactly correct - there will be a great loss of information (notably the full individual sequence data itself, as only the coordinates + variation will remain, plus the data will be in BED6, which means no splicing, just global coordinates - you won't want this for RNA-seq).
Since you are examining RNA-seq data, I also wanted to remind you of the choice to use Trackster for visualization (top Galaxy menu -> "Visualization"). A local or custom reference genome can be used, histogram or full read display is possible, and deeply sequenced areas can be "unpacked" in stages by using the visualization controls (really nice). Other features such as drag and drop composite track creation, new datasets from selects within Trackster (to feed more analysis), strand coloring, etc. are fairly recent enhancements (see: http://wiki.g2.bx.psu.edu/DevNewsBriefs/2012_03_12#Galaxy_Track_Browser_.28G... http://wiki.g2.bx.psu.edu/DevNewsBriefs/2012_05_11#Galaxy_Track_Browser_.28G...). More undocumented (so far!) features are added all the time, so good to experiment. A new paper is in the works that will cover the details. And there will be tutorials, screencasts, etc. later on, but for now during the active development cycle, just using it is best and is pretty intuitive. We're interested in what you like or wish was added (this is true to everyone reading this!). No promises, but is good to get feedback!
If there is a particular UCSC track (or another source mapped to same reference genome) that you wish to compare your data against, it could be imported into Galaxy and added to your Trackster visualization session. There are no known limits on data size/depth - only the general Galaxy dataset file size limit of 50G (applies to all datasets) - although I suppose an extreme corner case could probably be devised/uncovered. The point is that a routine RNA-seq experiment shouldn't present a problem. Special tuning to speed up and handle depth has been a big area of recent development activity. If you do run into issues with Trackster, feel free to ask questions (I'll probably ask you to send a share link to the visualization on the pubic main Galaxy instance and bring Jeremy into the discussion if the solution is not clear).
Please let us know if you need more help,
Jen
On 5/29/12 11:57 AM, Fowler, Trent wrote:
Hello,
I am attempting to run accepted hit data from Tophat output into the UCSC Genome Web Browser for visualization of sequencing hits in specific genes. However, the BAM files yield tiles and are too large to present through the browser. Is there a better file format to convert to that would allow better visualization such as histograms?
From word of mouth, I have been told to convert BAMs to BEDs and put BED files through the browser. However, I notice that Galaxy does not have an option for this and the oft used BEDtools appears to involve writing code, which is above my computer abilities.
Any tips or solutions on how to obtain histograms from sequencing data would be very welcome.
Thanks
Trent Fowler
___________________________________________________________
The Galaxy User list should be used for the discussion of
Galaxy analysis and other features on the public server
at usegalaxy.org. Please keep all replies on the list by
using"reply all" in your mail client. For discussion of
local Galaxy instances and the Galaxy source code, please
use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists,
please use the interface at:
--
Jennifer Jackson
-- Jennifer Jackson http://galaxyproject.org
{kind=link}
{kind=link}
participants (1)
-
 Jennifer Jackson
Jennifer Jackson