Create an index with samtools within a pipeline?

6 Nov
2012
6 Nov
'12
3:43 p.m.
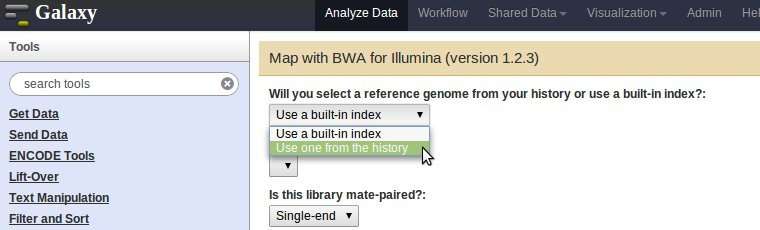
I want to make a pipeline that grabs a reference genome, indexes it, then aligns reads to it via bwa. The bwa tool has a menu option for using an index "from the history" (see attached image) but I don't see any option within the bwa wrappers to actually create an index on the fly. This is also true for samtools and other aligners. The following link describes the conf/loc files required to add indexes to the system: http://wiki.galaxyproject.org/Admin/NGS%20Local%20Setup But how can they be created dynamically in the course of a pipeline run instead of preconfigured in this way? Regards - Joshua
{kind=link}
4957
Age (days ago)
4957
Last active (days ago)
0 comments
1 participants
participants (1)
-
 Joshua Orvis
Joshua Orvis