
I am new to the NGS analysis. I need help to solve this problem. As shown in my previous emial/question shown below, I have some paired-end datasets at FASTQ format, and I have problem to split each of these datasets into two datasets (one forward and one reverse). Jennifer instructed me to assign the datatype to be fastqsanger first and then run 'Manipulate FASTQ'. I have two questions: 1) Now that the datasets were already split into forward and reverse reads when extracted in FASTQ format from the SRA, can I use them just as single end data? 2) If I do need to split each dataset into two datasets, how should I choose the settings when I run "Manipulte FASTQ"? Thanks. Jianguang ///////////////////////// On 8/10/12 7:21 AM, Du, Jianguang wrote:
I have problem to split a paired-end FASTQ dataset into two separate datasets. In order to explain the problem clearly, I list the detail of what I did with my dataset:
Step 1) My aim is to compare datasets for the differential alternative splicing. I downloaded paired-end datasets at FASTQ format from SRA of NCBI as original data.
Below is part of my paired-end FASTQ dataset that I downloaed from SRA of NCBI, Does this dataset look OK?
@SRR192532.1.1 HWI-EAS269:1:4:655:110.1 length=35 GTTTTCTGAGTGAGAAAAGGTGTGTTTGGAGTTTG +SRR192532.1.1 HWI-EAS269:1:4:655:110.1 length=35 I28II;II*2/<5:++,(..*943F@I.('+.35' @SRR192532.1.2 HWI-EAS269:1:4:655:110.2 length=35 AAAGATGTTAGTGTTTTATACGGAAAGGATATCTC +SRR192532.1.2 HWI-EAS269:1:4:655:110.2 length=35 9+*9+7@?F1206,IGI+D122&/0++-.>+6/@?
Step 2) Then I performed FASTQ groomer at setting as follows:
a) Input FASTQ quality scores type: Illumina 1.3-1.7
b)Advanced Options: Hide Advanced Options.
Did I choose the right setting for FASTQ groomer? Should I use Advanced Options? If yes, what is the setting for Advances Options?
Below is part of groomed dataset:
@SRR192532.1.1 HWI-EAS269:1:4:655:110.1 length=35 GTTTTCTGAGTGAGAAAAGGTGTGTTTGGAGTTTG +SRR192532.1.1 HWI-EAS269:1:4:655:110.1 length=35 *!!**!**!!!!!!!!!!!!!!!!'!*!!!!!!!! @SRR192532.1.2 HWI-EAS269:1:4:655:110.2 length=35 AAAGATGTTAGTGTTTTATACGGAAAGGATATCTC +SRR192532.1.2 HWI-EAS269:1:4:655:110.2 length=35 !!!!!!!!'!!!!!*(*!%!!!!!!!!!!!!!!!!
Does the groomed data look right? Is number represnting the member of a pair correct? Here they are ".1" and ".2", should they be "/1" and "/2"?
Step 3) Then I ran FASTQ splitter with the groomed files. There is not setting for the splitter. I chose the right groomed file and then click "Excute". Below is the description of the splitted dataset:
empty format:fastqsanger, database:hg19 Info: Split 0 of 15277248 reads (0.00%).
Please help me dela with this problem.
Thanks.
Jianguang Du

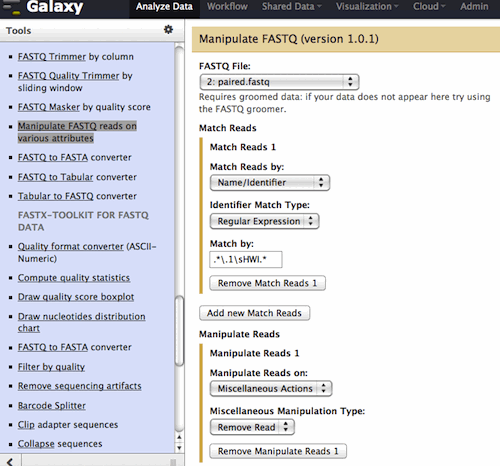
Hi Jianguang, I took a screenshot to simplify the instructions. Please see the attached. The tool is: 1 - filtering for a match against the identifier with a regular expression 2 - removing those matched reads, leaving the remainder Run twice, once with each regular expression. Remember that the reverse of the match will be in the output. .*\.1\sHWI.* <- this is in the attached screenshot .*\.2\sHWI.* Others expressions would work, these are just examples that you can use right now, for your exact data. I tried to not be overly cryptic so this could help as a base for future queries. @SRR192532.1.1 HWI-EAS269:1:4:655:110.1 length=35 ^^^^^^ I am matching the sequences where the ^^ are: at the end of the identifier, the first space, and the start of the description. The link on the tool form to the regular expression help is a good one to aid with understanding how/why this works. Hopefully this helps! Jen Galaxy team On 8/10/12 12:43 PM, Du, Jianguang wrote:
I am new to the NGS analysis. I need help to solve this problem.
As shown in my previous emial/question shown below, I have some paired-end datasets at FASTQ format, and I have problem to split each of these datasets into two datasets (one forward and one reverse).
Jennifer instructed me to assign the datatype to be fastqsanger first and then run 'Manipulate FASTQ'.
I have two questions:
1) Now that the datasets were already split into forward and reverse reads when extracted in FASTQ format from the SRA, can I use them just as single end data?
2) If I do need to split each dataset into two datasets, how should I choose the settings when I run "Manipulte FASTQ"?
Thanks.
Jianguang
-- Jennifer Jackson http://galaxyproject.org
{kind=link}
participants (2)
-
 Du, Jianguang
Du, Jianguang -
 Jennifer Jackson
Jennifer Jackson