
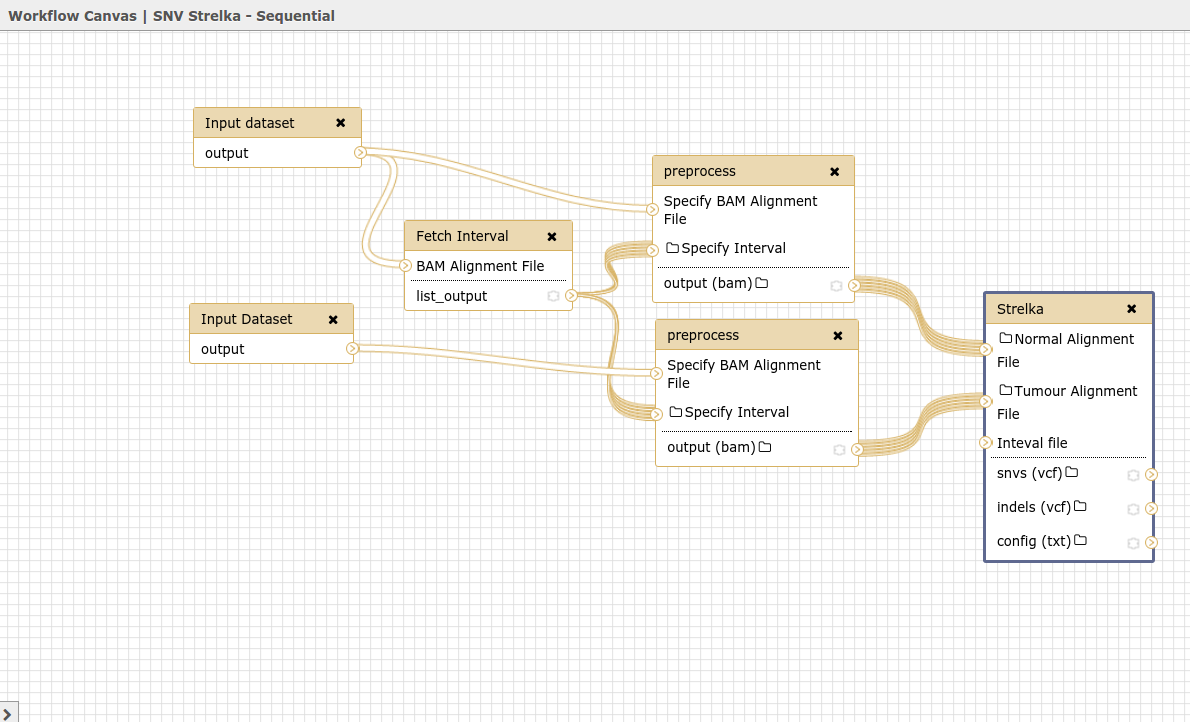
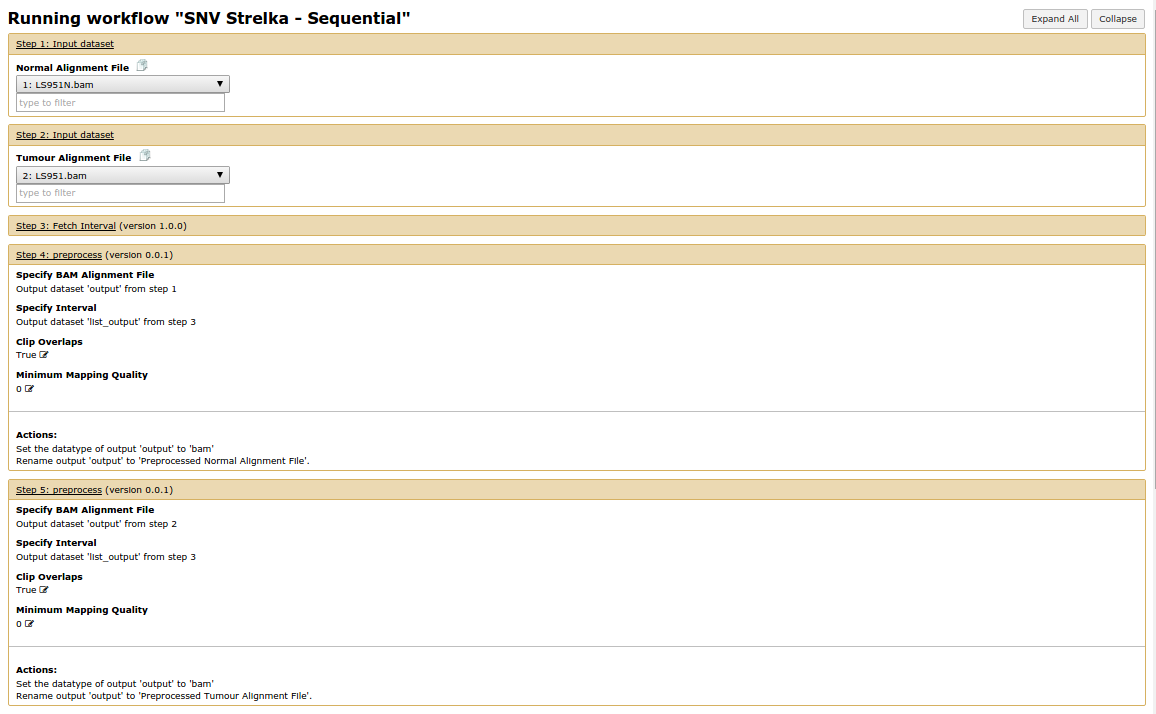
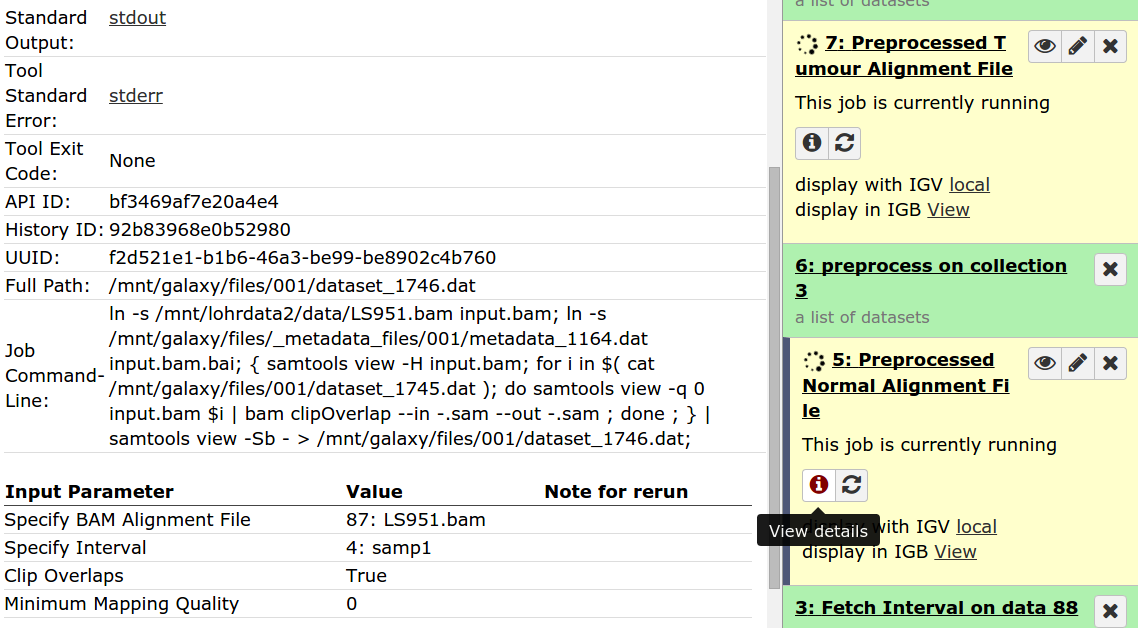
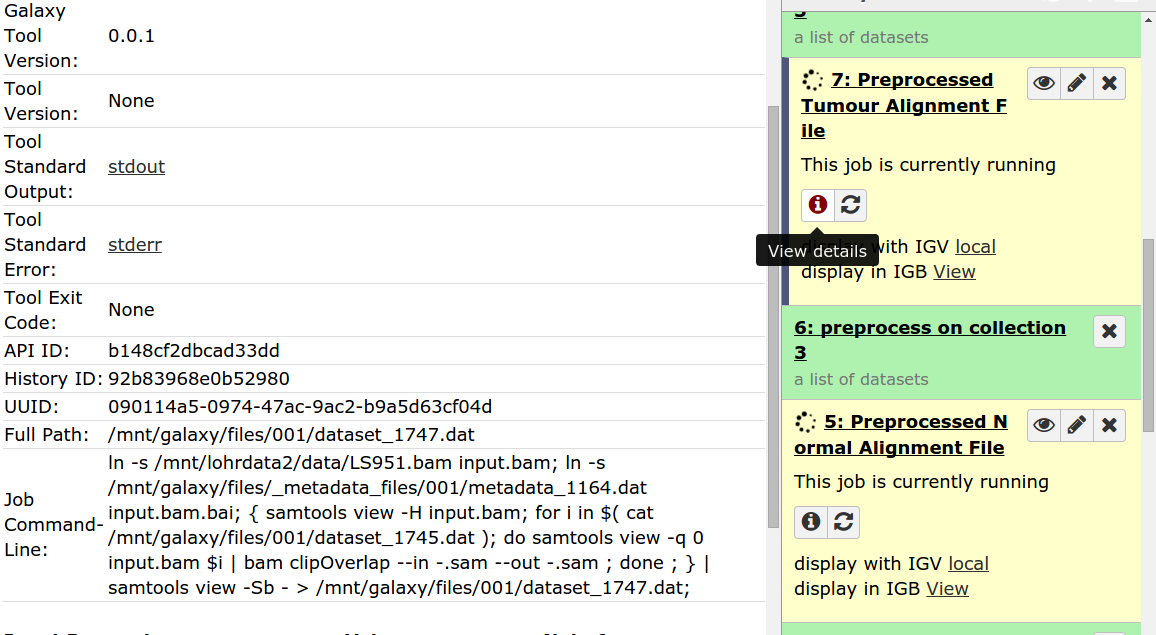
Hello Galaxy Dev, Consider the workflow found in 'Strelka_Workflow_Sequential' image Here it is in a different view 'Strelka_Sequential_Workflow_Different_View' Notice That the first input dataset links to the fetch_interval tool and the first preprocess tool. And the second input dataset links only to the second preprocess tool. When you run the workflow, what actually happens is the first input dataset goes to the fetch_interval tools and the second input dataset goes to both preprocessing tools. (Look at final two images). They have essentially executed the same tool twice. Where it should be an execution with a normal file and an execution with a tumour file. Why is this happening? What can I do to fix this? Thanks, Marco Albuquerque
{kind=link}
{kind=link}
{kind=link}
{kind=link}