Tool shed environment dependency - HOW TO do this?

Hi, I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick. I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is: * I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved. Thanks! Pieter Lukasse Wageningen UR, Plant Research International Department of Bioinformatics (Bioscience) Wageningen Campus, Building 107, Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands T: +31-317481122; M: +31-628189540; skype: pieter.lukasse.wur http://www.pri.wur.nl<http://www.pri.wur.nl/>

Hi Pieter, Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ... Hope this helps, Bjoern P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse Wageningen UR, Plant Research International Department of Bioinformatics (Bioscience) Wageningen Campus, Building 107, Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands T: +31-317481122; M: +31-628189540; skype: pieter.lukasse.wur http://www.pri.wur.nl<http://www.pri.wur.nl/>
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
Hi Bjoern, Thanks for this reply. I will have to try referring to the R_ROOT_DIR from another tool_dependency. If it doesn't work I'll come back ;) Regards, Pieter -----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ... Hope this helps, Bjoern P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse Wageningen UR, Plant Research International Department of Bioinformatics (Bioscience) Wageningen Campus, Building 107, Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands T: +31-317481122; M: +31-628189540; skype: pieter.lukasse.wur http://www.pri.wur.nl<http://www.pri.wur.nl/>
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
Hi Bjoern, I tried your suggestion, but it didn't work. So now I have split up the R part from the Bioconductor part (blue and yellow below, respectively). But the problem is that the <install> part executes before the <package> part is finished and the $R_ROOT_DIR reference also doesn't seem to work as I get the following error : " /bin/sh: 1: /bin/Rscript: not found " . This R_ROOT_DIR is set by the <package> script in my other tool_dependency.xml but is not visible in the tool_dependency.xml below. But maybe I misunderstood what you meant by "You can now access R_ROOT_DIR from any other tool_dependency file ..."? <tool_dependency> <!-- see also http://wiki.galaxyproject.org/ToolShedToolFeatures for syntax help --> <package name="R_bioc_metams" version="3.1.1"> <repository changeset_revision="0af753942e7b" name="prims_metabolomics_dependencies" owner="pieterlukasse" prior_installation_required="True" toolshed="http://testtoolshed.g2.bx.psu.edu" /> <install version="1.0"> <actions_group> <!-- the Bioconductor and metaMS part --> <action type="shell_command">wget -P $INSTALL_DIR http://testtoolshed.g2.bx.psu.edu/repos/pieterlukasse/prims_metabolomics/raw-file/tip/INSTALL.r</action> <action type="shell_command">$R_ROOT_DIR/bin/Rscript $INSTALL_DIR/INSTALL.r</action> </actions_group> </install> </package> <readme> This dependency: Ensures R 3.1.1 installation is triggered (via dependency). Ensures Bioconductor 3.0 and package metaMS, multtest and snow are installed. </readme> </tool_dependency> Thanks, Pieter -----Original Message----- From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Lukasse, Pieter Sent: donderdag 6 november 2014 10:02 To: 'Björn Grüning'; galaxy-dev@lists.bx.psu.edu; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Bjoern, Thanks for this reply. I will have to try referring to the R_ROOT_DIR from another tool_dependency. If it doesn't work I'll come back ;) Regards, Pieter -----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu<mailto:galaxy-dev@lists.bx.psu.edu>; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ... Hope this helps, Bjoern P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122;
M: +31-628189540;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/<http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/

Hi Pieter, you should probably add: <action type="set_environment_for_install"> <repository name="prims_metabolomics_r_dependencies" owner="pieterlukasse"> <package name="R_bioc_metams" version="3.1.1" /> </repository> </action> before using the R_ROOT_DIR environment variable. Best, Nicola Il 2014-11-06 11:28 Lukasse, Pieter ha scritto:
Hi Bjoern,
I tried your suggestion, but it didn't work. So now I have split up the R part from the Bioconductor part (blue and yellow below, respectively). But the problem is that the <install> part executes before the <package> part is finished and the $R_ROOT_DIR reference also doesn't seem to work as I get the following error : " /bin/sh: 1: /bin/Rscript: not found " . This R_ROOT_DIR is set by the <package> script in my other tool_dependency.xml but is not visible in the tool_dependency.xml below. But maybe I misunderstood what you meant by "You can now access R_ROOT_DIR from any other tool_dependency file ..."?
<tool_dependency>
<!-- see also http://wiki.galaxyproject.org/ToolShedToolFeatures for syntax help
-->
<package name="R_bioc_metams" version="3.1.1">
<repository changeset_revision="0af753942e7b" name="prims_metabolomics_dependencies" owner="pieterlukasse" prior_installation_required="True" toolshed="http://testtoolshed.g2.bx.psu.edu" />
<install version="1.0">
<actions_group>
<!-- the Bioconductor and metaMS part -->
<action type="shell_command">wget -P $INSTALL_DIR
<action type="shell_command">$R_ROOT_DIR/bin/Rscript $INSTALL_DIR/INSTALL.r</action>
</actions_group>
</install>
</package>
<readme>
This dependency:
Ensures R 3.1.1 installation is triggered (via dependency).
Ensures Bioconductor 3.0 and package metaMS, multtest and snow are installed.
</readme>
</tool_dependency>
Thanks,
Pieter
-----Original Message----- From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Lukasse, Pieter Sent: donderdag 6 november 2014 10:02 To: 'Björn Grüning'; galaxy-dev@lists.bx.psu.edu; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Bjoern,
Thanks for this reply. I will have to try referring to the R_ROOT_DIR from another tool_dependency. If it doesn't work I'll come back ;)
Regards,
Pieter
-----Original Message-----
From: Björn Grüning [mailto:bjoern.gruening@gmail.com [1]]
Sent: donderdag 30 oktober 2014 11:25
To: galaxy-dev@lists.bx.psu.edu [2]; Lukasse, Pieter; Dave Bouvier
Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at
https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... [3]
You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ...
Hope this helps,
Bjoern
P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122;
M: +31-628189540;
skype: pieter.lukasse.wur
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
http://galaxyproject.org/search/mailinglists/ [8]
Links: ------ [1] mailto:bjoern.gruening@gmail.com [2] mailto:galaxy-dev@lists.bx.psu.edu [3]
https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... [4] http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/ [5] http://lists.bx.psu.edu/ [6] http://galaxyproject.org/search/mailinglists/ [7] http://lists.bx.psu.edu/ [8] http://galaxyproject.org/search/mailinglists/
I agree with Nicola here! :) Am 06.11.2014 um 17:48 schrieb Nicola Soranzo:
Hi Pieter, you should probably add:
<action type="set_environment_for_install"> <repository name="prims_metabolomics_r_dependencies" owner="pieterlukasse"> <package name="R_bioc_metams" version="3.1.1" /> </repository> </action>
before using the R_ROOT_DIR environment variable.
Best, Nicola
Il 2014-11-06 11:28 Lukasse, Pieter ha scritto:
Hi Bjoern,
I tried your suggestion, but it didn't work. So now I have split up the R part from the Bioconductor part (blue and yellow below, respectively). But the problem is that the <install> part executes before the <package> part is finished and the $R_ROOT_DIR reference also doesn't seem to work as I get the following error : " /bin/sh: 1: /bin/Rscript: not found " . This R_ROOT_DIR is set by the <package> script in my other tool_dependency.xml but is not visible in the tool_dependency.xml below. But maybe I misunderstood what you meant by "You can now access R_ROOT_DIR from any other tool_dependency file ..."?
<tool_dependency>
<!-- see also http://wiki.galaxyproject.org/ToolShedToolFeatures for syntax help
-->
<package name="R_bioc_metams" version="3.1.1">
<repository changeset_revision="0af753942e7b" name="prims_metabolomics_dependencies" owner="pieterlukasse" prior_installation_required="True" toolshed="http://testtoolshed.g2.bx.psu.edu" />
<install version="1.0">
<actions_group>
<!-- the Bioconductor and metaMS part -->
<action type="shell_command">wget -P $INSTALL_DIR
<action type="shell_command">$R_ROOT_DIR/bin/Rscript $INSTALL_DIR/INSTALL.r</action>
</actions_group>
</install>
</package>
<readme>
This dependency:
Ensures R 3.1.1 installation is triggered (via dependency).
Ensures Bioconductor 3.0 and package metaMS, multtest and snow are installed.
</readme>
</tool_dependency>
Thanks,
Pieter
-----Original Message----- From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Lukasse, Pieter Sent: donderdag 6 november 2014 10:02 To: 'Björn Grüning'; galaxy-dev@lists.bx.psu.edu; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Bjoern,
Thanks for this reply. I will have to try referring to the R_ROOT_DIR from another tool_dependency. If it doesn't work I'll come back ;)
Regards,
Pieter
-----Original Message-----
From: Björn Grüning [mailto:bjoern.gruening@gmail.com [1]]
Sent: donderdag 30 oktober 2014 11:25
To: galaxy-dev@lists.bx.psu.edu [2]; Lukasse, Pieter; Dave Bouvier
Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at
https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_...
[3]
You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ...
Hope this helps,
Bjoern
P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122;
M: +31-628189540;
skype: pieter.lukasse.wur
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
http://galaxyproject.org/search/mailinglists/ [8]
Links: ------ [1] mailto:bjoern.gruening@gmail.com [2] mailto:galaxy-dev@lists.bx.psu.edu [3]
https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_...
[4] http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/ [5] http://lists.bx.psu.edu/ [6] http://galaxyproject.org/search/mailinglists/ [7] http://lists.bx.psu.edu/ [8] http://galaxyproject.org/search/mailinglists/
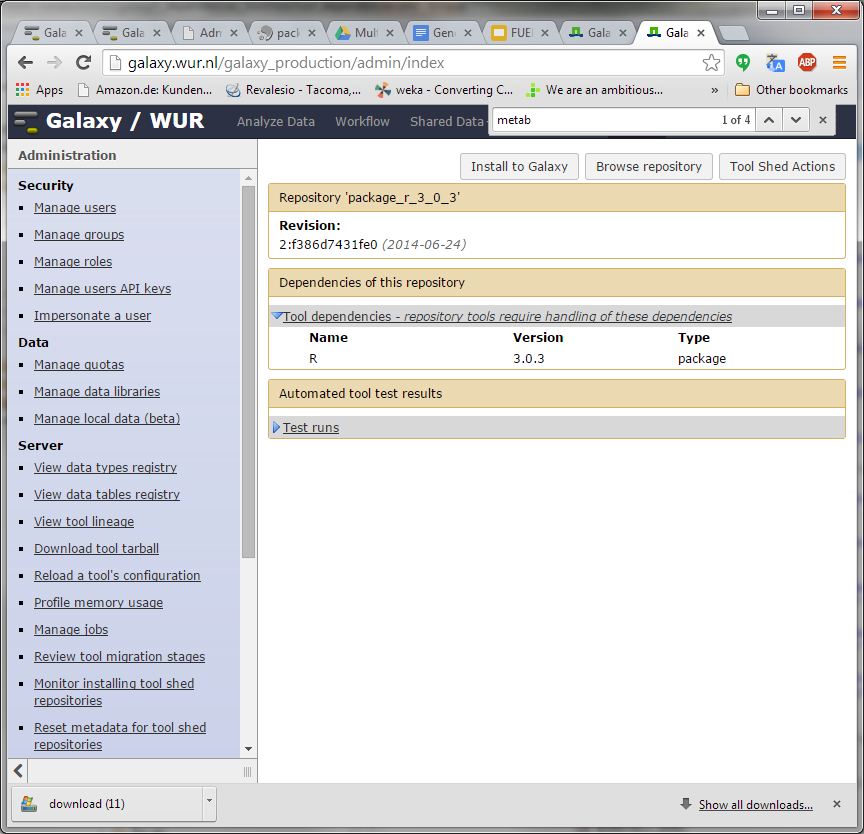
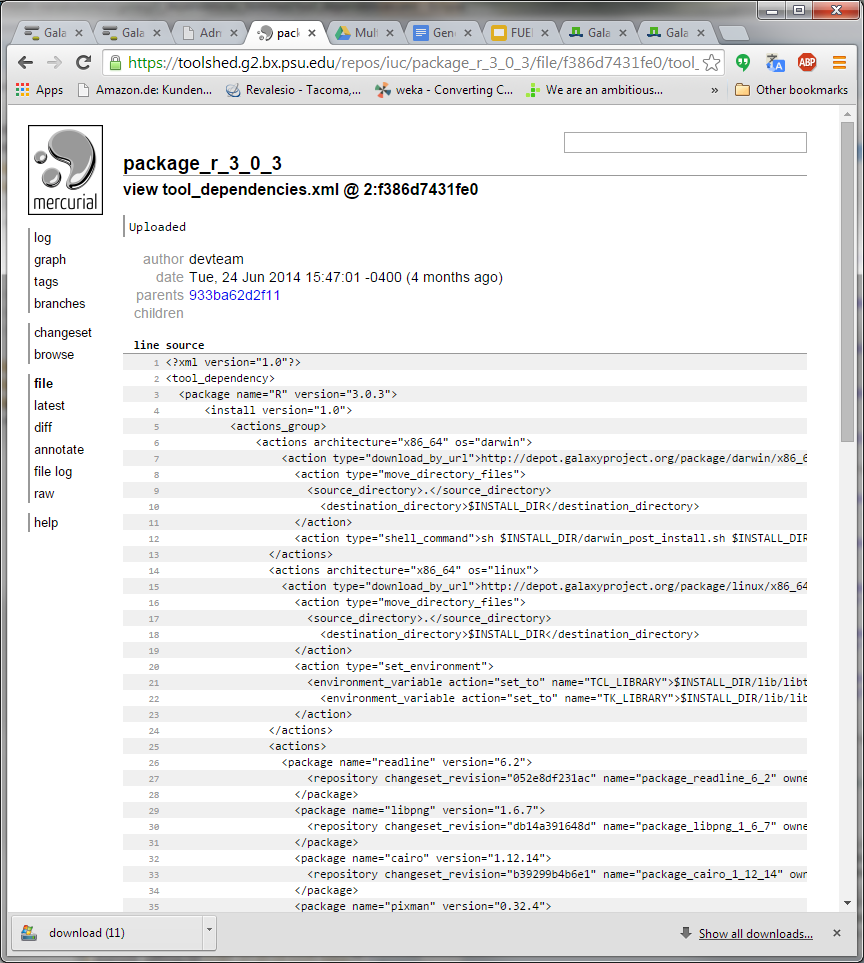
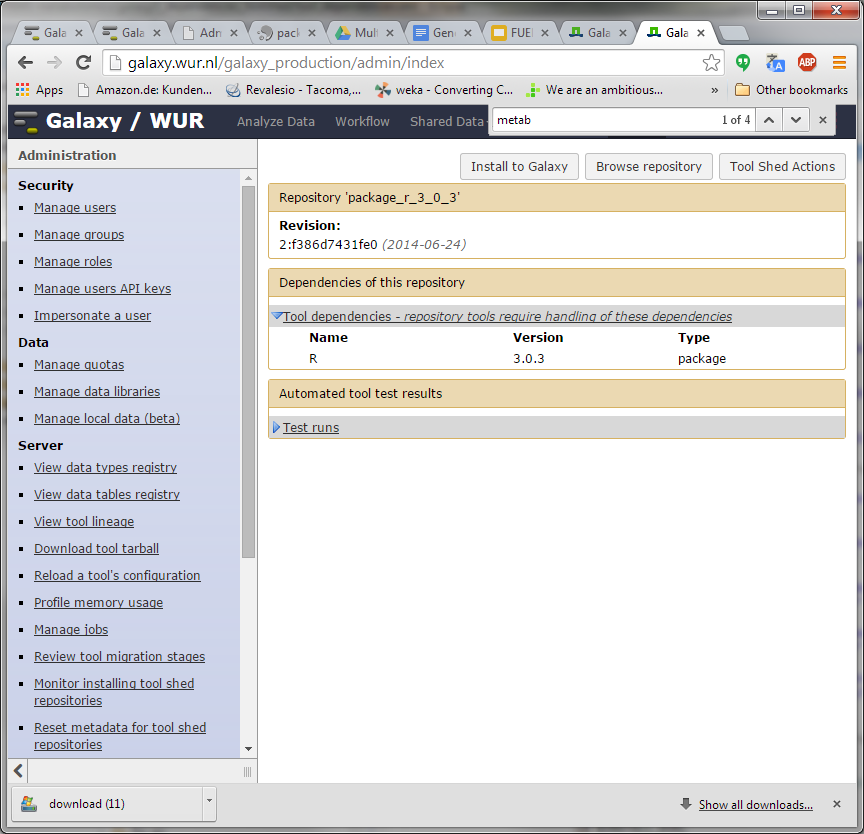
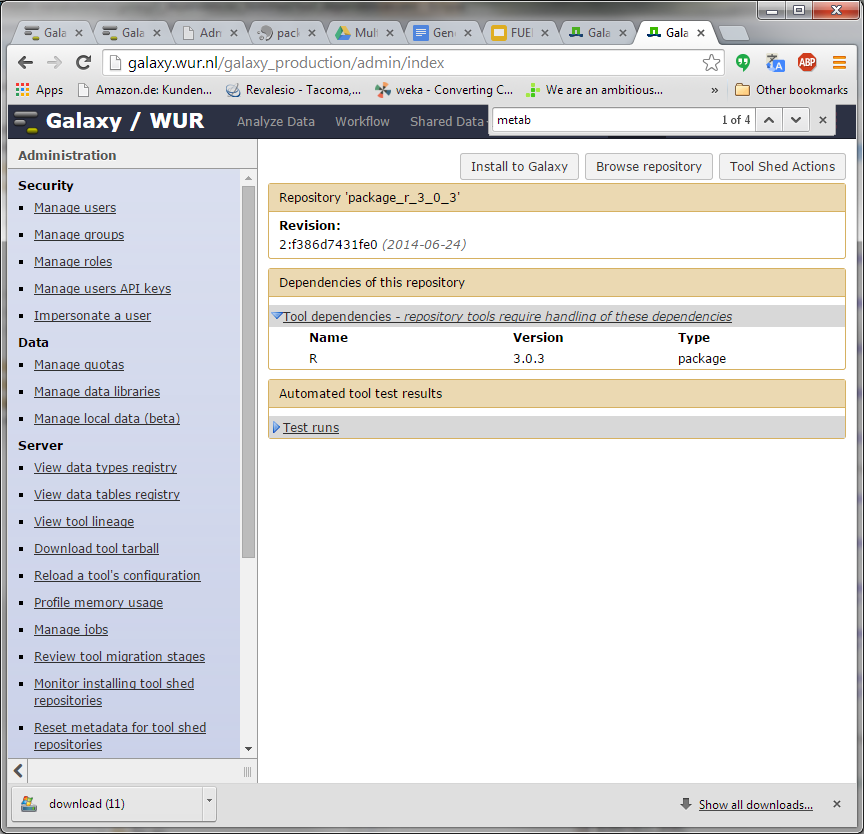
Hi Bjoern, Dave, I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works . It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all? Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org . So why the ./configure and make commands further down the XML? Thanks for your help! Pieter [cid:image001.png@01CFFE98.D1C9EF70] [cid:image002.png@01CFFE98.D1C9EF70] -----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ... Hope this helps, Bjoern P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122;
M: +31-628189540;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/<http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
{kind=link}
{kind=link}
Hi Pieter, the compilation and hence the dependencies on the libraries for compilation are a fallback mechanism. This recipe will try to fetch binaries, if this does not work, or if the admin reconfigured Galaxy ... we try to compile R by our own. Hope this helps, Bjoern 2014-11-12 16:57 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl>:
Hi Bjoern, Dave,
I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works .
It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all?
Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org . So why the ./configure and make commands further down the XML?
Thanks for your help!
Pieter
-----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_...
You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ...
Hope this helps,
Bjoern
P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122;
M: +31-628189540;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/ <http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
{kind=link}
{kind=link}
Ok, I didn’t know that...but it does explain the behavior we are observing. So the <actions_group> tag is a kind of switch/try mechanism where the next applicable <actions> part is only executed if the previous one fails? Is that how I should read it? So my script is based on the last part, which actually tries to compile R with cairo....and unfortunately this is not working. But maybe this was not detected by others because as you wrote, the compilation part is only a fall back (and probably will not be executed in practice)...? Thanks, Pieter. From: bjoern.gruening@googlemail.com [mailto:bjoern.gruening@gmail.com] Sent: woensdag 12 november 2014 17:01 To: Lukasse, Pieter Cc: galaxy-dev@lists.bx.psu.edu; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, the compilation and hence the dependencies on the libraries for compilation are a fallback mechanism. This recipe will try to fetch binaries, if this does not work, or if the admin reconfigured Galaxy ... we try to compile R by our own. Hope this helps, Bjoern 2014-11-12 16:57 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl<mailto:pieter.lukasse@wur.nl>>: Hi Bjoern, Dave, I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works . It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all? Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org<http://depot.galaxy.org> . So why the ./configure and make commands further down the XML? Thanks for your help! Pieter [cid:image001.png@01CFFE9B.473A68F0] [cid:image002.png@01CFFE9B.473A68F0] -----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com<mailto:bjoern.gruening@gmail.com>] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu<mailto:galaxy-dev@lists.bx.psu.edu>; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ... Hope this helps, Bjoern P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122<tel:%2B31-317481122>;
M: +31-628189540<tel:%2B31-628189540>;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/<http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
{kind=link}
{kind=link}
Yes, this is correct. Do you really need to compile it by yourself? This is really a pain, I tried it during the R2 lifetime and this wasn't fun. 2014-11-12 17:10 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl>:
Ok, I didn’t know that...but it does explain the behavior we are observing. So the <actions_group> tag is a kind of switch/try mechanism where the next applicable <actions> part is only executed if the previous one fails? Is that how I should read it?
So my script is based on the last part, which actually tries to compile R with cairo....and unfortunately this is not working. But maybe this was not detected by others because as you wrote, the compilation part is only a fall back (and probably will not be executed in practice)...?
Thanks,
Pieter.
*From:* bjoern.gruening@googlemail.com [mailto:bjoern.gruening@gmail.com] *Sent:* woensdag 12 november 2014 17:01 *To:* Lukasse, Pieter *Cc:* galaxy-dev@lists.bx.psu.edu; Dave Bouvier
*Subject:* Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
the compilation and hence the dependencies on the libraries for compilation are a fallback mechanism. This recipe will try to fetch binaries, if this does not work, or if the admin reconfigured Galaxy ... we try to compile R by our own.
Hope this helps,
Bjoern
2014-11-12 16:57 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl>:
Hi Bjoern, Dave,
I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works .
It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all?
Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org . So why the ./configure and make commands further down the XML?
Thanks for your help!
Pieter
-----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_...
You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ...
Hope this helps,
Bjoern
P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122;
M: +31-628189540;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/ <http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
{kind=link}
{kind=link}
Indeed...painful and slow :( What I need is R 3.1.1 or later...so if I can get a pre-compiled package in toolshed that works I’m also happy! I understand Dave manages this process? @Dave: can you help me? Thanks, Pieter From: bjoern.gruening@googlemail.com [mailto:bjoern.gruening@gmail.com] Sent: woensdag 12 november 2014 17:18 To: Lukasse, Pieter Cc: galaxy-dev@lists.bx.psu.edu; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Yes, this is correct. Do you really need to compile it by yourself? This is really a pain, I tried it during the R2 lifetime and this wasn't fun. 2014-11-12 17:10 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl<mailto:pieter.lukasse@wur.nl>>: Ok, I didn’t know that...but it does explain the behavior we are observing. So the <actions_group> tag is a kind of switch/try mechanism where the next applicable <actions> part is only executed if the previous one fails? Is that how I should read it? So my script is based on the last part, which actually tries to compile R with cairo....and unfortunately this is not working. But maybe this was not detected by others because as you wrote, the compilation part is only a fall back (and probably will not be executed in practice)...? Thanks, Pieter. From: bjoern.gruening@googlemail.com<mailto:bjoern.gruening@googlemail.com> [mailto:bjoern.gruening@gmail.com<mailto:bjoern.gruening@gmail.com>] Sent: woensdag 12 november 2014 17:01 To: Lukasse, Pieter Cc: galaxy-dev@lists.bx.psu.edu<mailto:galaxy-dev@lists.bx.psu.edu>; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, the compilation and hence the dependencies on the libraries for compilation are a fallback mechanism. This recipe will try to fetch binaries, if this does not work, or if the admin reconfigured Galaxy ... we try to compile R by our own. Hope this helps, Bjoern 2014-11-12 16:57 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl<mailto:pieter.lukasse@wur.nl>>: Hi Bjoern, Dave, I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works . It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all? Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org<http://depot.galaxy.org> . So why the ./configure and make commands further down the XML? Thanks for your help! Pieter [cid:image001.png@01CFFE9D.A5EB3710] [cid:image002.png@01CFFE9D.A5EB3710] -----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com<mailto:bjoern.gruening@gmail.com>] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu<mailto:galaxy-dev@lists.bx.psu.edu>; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ... Hope this helps, Bjoern P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122<tel:%2B31-317481122>;
M: +31-628189540<tel:%2B31-628189540>;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/<http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
{kind=link}
{kind=link}

Pieter, I am in fact planning on compiling R 3.1.1 for depot.galaxyproject in the near future. --Dave B. On 11/12/2014 11:25 AM, Lukasse, Pieter wrote:
Indeed...painful and slow :(
What I need is R 3.1.1 or later...so if I can get a pre-compiled package in toolshed that works I’m also happy! I understand Dave manages this process?
@Dave: can you help me?
Thanks,
Pieter
*From:*bjoern.gruening@googlemail.com [mailto:bjoern.gruening@gmail.com] *Sent:* woensdag 12 november 2014 17:18 *To:* Lukasse, Pieter *Cc:* galaxy-dev@lists.bx.psu.edu; Dave Bouvier *Subject:* Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Yes, this is correct. Do you really need to compile it by yourself? This is really a pain, I tried it during the R2 lifetime and this wasn't fun.
2014-11-12 17:10 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl <mailto:pieter.lukasse@wur.nl>>:
Ok, I didn’t know that...but it does explain the behavior we are observing. So the <actions_group> tag is a kind of switch/try mechanism where the next applicable <actions> part is only executed if the previous one fails? Is that how I should read it?
So my script is based on the last part, which actually tries to compile R with cairo....and unfortunately this is not working. But maybe this was not detected by others because as you wrote, the compilation part is only a fall back (and probably will not be executed in practice)...?
Thanks,
Pieter.
*From:*bjoern.gruening@googlemail.com <mailto:bjoern.gruening@googlemail.com> [mailto:bjoern.gruening@gmail.com <mailto:bjoern.gruening@gmail.com>] *Sent:* woensdag 12 november 2014 17:01 *To:* Lukasse, Pieter *Cc:* galaxy-dev@lists.bx.psu.edu <mailto:galaxy-dev@lists.bx.psu.edu>; Dave Bouvier
*Subject:* Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
the compilation and hence the dependencies on the libraries for compilation are a fallback mechanism. This recipe will try to fetch binaries, if this does not work, or if the admin reconfigured Galaxy ... we try to compile R by our own.
Hope this helps,
Bjoern
2014-11-12 16:57 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl <mailto:pieter.lukasse@wur.nl>>:
Hi Bjoern, Dave,
I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works .
It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all?
Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org <http://depot.galaxy.org> . So why the ./configure and make commands further down the XML?
Thanks for your help!
Pieter
-----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com <mailto:bjoern.gruening@gmail.com>] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu <mailto:galaxy-dev@lists.bx.psu.edu>; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_...
You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ...
Hope this helps,
Bjoern
P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122 <tel:%2B31-317481122>;
M: +31-628189540 <tel:%2B31-628189540>;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/ <http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
Ah, great! Please let me know once I can use it. Thanks, Pieter -----Original Message----- From: Dave Bouvier [mailto:dave@bx.psu.edu] Sent: woensdag 12 november 2014 17:27 To: Lukasse, Pieter; 'bjoern.gruening@googlemail.com' Cc: galaxy-dev@lists.bx.psu.edu Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Pieter, I am in fact planning on compiling R 3.1.1 for depot.galaxyproject in the near future. --Dave B. On 11/12/2014 11:25 AM, Lukasse, Pieter wrote:
Indeed...painful and slow :(
What I need is R 3.1.1 or later...so if I can get a pre-compiled package in toolshed that works I’m also happy! I understand Dave manages this process?
@Dave: can you help me?
Thanks,
Pieter
*From:*bjoern.gruening@googlemail.com [mailto:bjoern.gruening@gmail.com] *Sent:* woensdag 12 november 2014 17:18 *To:* Lukasse, Pieter *Cc:* galaxy-dev@lists.bx.psu.edu; Dave Bouvier *Subject:* Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Yes, this is correct. Do you really need to compile it by yourself? This is really a pain, I tried it during the R2 lifetime and this wasn't fun.
2014-11-12 17:10 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl <mailto:pieter.lukasse@wur.nl>>:
Ok, I didn’t know that...but it does explain the behavior we are observing. So the <actions_group> tag is a kind of switch/try mechanism where the next applicable <actions> part is only executed if the previous one fails? Is that how I should read it?
So my script is based on the last part, which actually tries to compile R with cairo....and unfortunately this is not working. But maybe this was not detected by others because as you wrote, the compilation part is only a fall back (and probably will not be executed in practice)...?
Thanks,
Pieter.
*From:*bjoern.gruening@googlemail.com <mailto:bjoern.gruening@googlemail.com> [mailto:bjoern.gruening@gmail.com <mailto:bjoern.gruening@gmail.com>] *Sent:* woensdag 12 november 2014 17:01 *To:* Lukasse, Pieter *Cc:* galaxy-dev@lists.bx.psu.edu <mailto:galaxy-dev@lists.bx.psu.edu>; Dave Bouvier
*Subject:* Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
the compilation and hence the dependencies on the libraries for compilation are a fallback mechanism. This recipe will try to fetch binaries, if this does not work, or if the admin reconfigured Galaxy ... we try to compile R by our own.
Hope this helps,
Bjoern
2014-11-12 16:57 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl <mailto:pieter.lukasse@wur.nl>>:
Hi Bjoern, Dave,
I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works .
It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all?
Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org <http://depot.galaxy.org> . So why the ./configure and make commands further down the XML?
Thanks for your help!
Pieter
-----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com <mailto:bjoern.gruening@gmail.com>] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu <mailto:galaxy-dev@lists.bx.psu.edu>; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this?
Hi Pieter,
Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_ r_3_0_3/tool_dependencies.xml
You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ...
Hope this helps,
Bjoern
P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122 <tel:%2B31-317481122>;
M: +31-628189540 <tel:%2B31-628189540>;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/ <http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
Indeed...painful and slow :( What I need is R 3.1.1 or later...so if I can get a pre-compiled package in toolshed that works I’m also happy! I understand Dave manages this process? @Dave: can you help me? Thanks, Pieter From: bjoern.gruening@googlemail.com [mailto:bjoern.gruening@gmail.com] Sent: woensdag 12 november 2014 17:18 To: Lukasse, Pieter Cc: galaxy-dev@lists.bx.psu.edu; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Yes, this is correct. Do you really need to compile it by yourself? This is really a pain, I tried it during the R2 lifetime and this wasn't fun. 2014-11-12 17:10 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl<mailto:pieter.lukasse@wur.nl>>: Ok, I didn’t know that...but it does explain the behavior we are observing. So the <actions_group> tag is a kind of switch/try mechanism where the next applicable <actions> part is only executed if the previous one fails? Is that how I should read it? So my script is based on the last part, which actually tries to compile R with cairo....and unfortunately this is not working. But maybe this was not detected by others because as you wrote, the compilation part is only a fall back (and probably will not be executed in practice)...? Thanks, Pieter. From: bjoern.gruening@googlemail.com<mailto:bjoern.gruening@googlemail.com> [mailto:bjoern.gruening@gmail.com<mailto:bjoern.gruening@gmail.com>] Sent: woensdag 12 november 2014 17:01 To: Lukasse, Pieter Cc: galaxy-dev@lists.bx.psu.edu<mailto:galaxy-dev@lists.bx.psu.edu>; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, the compilation and hence the dependencies on the libraries for compilation are a fallback mechanism. This recipe will try to fetch binaries, if this does not work, or if the admin reconfigured Galaxy ... we try to compile R by our own. Hope this helps, Bjoern 2014-11-12 16:57 GMT+01:00 Lukasse, Pieter <pieter.lukasse@wur.nl<mailto:pieter.lukasse@wur.nl>>: Hi Bjoern, Dave, I have been trying to understand how https://toolshed.g2.bx.psu.edu/repos/iuc/package_r_3_0_3 works . It contains a number of package dependencies as shown in the XML below. However, none show up in the toolshed UI (next screenshot). Is this because the package entries (libpng, cairo, etc) are inside a <actions> tag while they should be outside of this tag? Could it be these are not executed after all? Furthermore, the installation actually seems to be a simple download of a pre-compiled R version from depot.galaxy.org<http://depot.galaxy.org> . So why the ./configure and make commands further down the XML? Thanks for your help! Pieter -----Original Message----- From: Björn Grüning [mailto:bjoern.gruening@gmail.com<mailto:bjoern.gruening@gmail.com>] Sent: donderdag 30 oktober 2014 11:25 To: galaxy-dev@lists.bx.psu.edu<mailto:galaxy-dev@lists.bx.psu.edu>; Lukasse, Pieter; Dave Bouvier Subject: Re: [galaxy-dev] Tool shed environment dependency - HOW TO do this? Hi Pieter, Am 30.10.2014 um 11:15 schrieb Lukasse, Pieter:
Hi,
I have a Bioconductor package which depends on R 3.1.1 , so I think I cannot use the "setup_r_environment" trick.
Why? Do you need a new release? The latest current release in the Tool Shed is R 3.1.0. If this is not enough we need to poke Dave to build a new package. But this way would be the preferred way.
I already got a tool_dependency.xml working that installs R 3.1.1 and the necessary Bioconductor packages using bioclite method (see attachment). Now my question is:
* I would like to split up this into two steps as I don't want to trigger the compilation of new R environment every time I when I need to just update the
Bioconductor package....the question is: how to do such things in general? How can I access the INSTALL_DIR of another tool from within another tool_dependency.xml? If I can do this, then my problem is solved.
If you really want to build your R packages by your own. Have a look at https://github.com/bgruening/galaxytools/blob/master/packages/package_r_3_0_... You will find an exhaustive installation instruction how to build R properly. Also note that we export a variable called R_ROOT_DIR that is set to $INSTALL_DIR. You can now access R_ROOT_DIR from any other tool_dependency file ... Hope this helps, Bjoern P.S. Do you mind asking this question on biostar again, I think this is a very nice question that can be of interest to a lot more people.
Thanks!
Pieter Lukasse
Wageningen UR, Plant Research International Department of
Bioinformatics (Bioscience) Wageningen Campus, Building 107,
Droevendaalsesteeg 1, 6708 PB, Wageningen, the Netherlands
T: +31-317481122<tel:%2B31-317481122>;
M: +31-628189540<tel:%2B31-628189540>;
skype: pieter.lukasse.wur
http://www.pri.wur.nl<http://www.pri.wur.nl/<http://www.pri.wur.nl%3chttp:/www.pri.wur.nl/>>
___________________________________________________________
Please keep all replies on the list by using "reply all"
in your mail client. To manage your subscriptions to this and other
Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
participants (5)
-
 bjoern.gruening@googlemail.com
bjoern.gruening@googlemail.com -
Björn Grüning
-
 Dave Bouvier
Dave Bouvier -
 Lukasse, Pieter
Lukasse, Pieter -
 Nicola Soranzo
Nicola Soranzo