version of SAM-to-BAM (CloudMap)

Dear there, When I run CloudMap EMS Variant Density Mapping workflow (takes VCF of heterozygous and homozygous variants to subtract) (imported from uploaded file) on local Galaxy instance, I got this [X] , when I wanted to edit the workflow. Also, I got this The following tools are beinge executed with a different version from what was available when this workflow was last saved because the previous version is no longer available for use on this galaxy instance. To upgrade your workflow and dismiss this message simply edit the workflow and re-save it to update the stored tool version. * toolshed.g2.bx.psu.edu/repos/devteam/sam_to_bam/sam_to_bam/1.1.2: using version '1.1.2' instead of version '1.1.3' indicated in this workflow. * ,when I wanted to run the workflow. I know it is the problem with version of SAM-to-BAM. But, when I searched the Tool sheds > Search and browse tool sheds, there is only version 1.1.4 for SAM-to-BAM. When I tried to edit the workflow from Workflow > ...> Edit, I did not find where could I change the version of SAM-to-BAM. When I clicked on SAM-to-BAM on the Workflow Canvas, it showed the version is 1.1.3 for it. Could you tell me which version do I need exactly, and how to figure out this? Thanks a lot! Best, Xiaofei

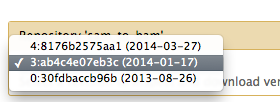
Hi Xiaofei, You should be able to install 1.1.3 by selecting revision 3 of sam_to_bam from the "Preview and install" page (Admin -> Search and browse tool sheds -> Galaxy main tool shed -> sam_to_bam -> Preview and install), and then clicking "Install to Galaxy." [image: Inline image 1] You can tell what version of the tool it'll install by looking at the "Valid tools" section of the "Contents of this repository" box. --nate On Thu, Apr 3, 2014 at 6:16 PM, Wang, Xiaofei <xfwang@ku.edu> wrote:
Dear there,
When I run CloudMap EMS Variant Density Mapping workflow (takes VCF of heterozygous and homozygous variants to subtract) (imported from uploaded file) on local Galaxy instance, I got this , when I wanted to edit the workflow. Also, I got this The following tools are beinge executed with a different version from what was available when this workflow was last saved because the previous version is no longer available for use on this galaxy instance. To upgrade your workflow and dismiss this message simply edit the workflow and re-save it to update the stored tool version.
- toolshed.g2.bx.psu.edu/repos/devteam/sam_to_bam/sam_to_bam/1.1.2: using version '1.1.2' instead of version '1.1.3' indicated in this workflow. - ,when I wanted to run the workflow.
I know it is the problem with version of SAM-to-BAM. But, when I searched the Tool sheds > Search and browse tool sheds, there is only version 1.1.4 for SAM-to-BAM. When I tried to edit the workflow from Workflow > ...> Edit, I did not find where could I change the version of SAM-to-BAM. When I clicked on SAM-to-BAM on the Workflow Canvas, it showed the version is 1.1.3 for it.
Could you tell me which version do I need exactly, and how to figure out this?
Thanks a lot!
Best,
Xiaofei
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
{kind=link}
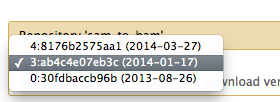
Yes, it is figured out when I downloaded the version of 1.1.2. I thought is no longer available. Thank you so much! Best, Xiaofei ________________________________ From: Nate Coraor [nate@bx.psu.edu] Sent: Friday, April 04, 2014 11:20 AM To: Wang, Xiaofei Cc: galaxy-dev@lists.bx.psu.edu Subject: Re: [galaxy-dev] version of SAM-to-BAM (CloudMap) Hi Xiaofei, You should be able to install 1.1.3 by selecting revision 3 of sam_to_bam from the "Preview and install" page (Admin -> Search and browse tool sheds -> Galaxy main tool shed -> sam_to_bam -> Preview and install), and then clicking "Install to Galaxy." [Inline image 1] You can tell what version of the tool it'll install by looking at the "Valid tools" section of the "Contents of this repository" box. --nate On Thu, Apr 3, 2014 at 6:16 PM, Wang, Xiaofei <xfwang@ku.edu<mailto:xfwang@ku.edu>> wrote: Dear there, When I run CloudMap EMS Variant Density Mapping workflow (takes VCF of heterozygous and homozygous variants to subtract) (imported from uploaded file) on local Galaxy instance, I got this [X] , when I wanted to edit the workflow. Also, I got this The following tools are beinge executed with a different version from what was available when this workflow was last saved because the previous version is no longer available for use on this galaxy instance. To upgrade your workflow and dismiss this message simply edit the workflow and re-save it to update the stored tool version. * toolshed.g2.bx.psu.edu/repos/devteam/sam_to_bam/sam_to_bam/1.1.2<http://toolshed.g2.bx.psu.edu/repos/devteam/sam_to_bam/sam_to_bam/1.1.2>: using version '1.1.2' instead of version '1.1.3' indicated in this workflow. * ,when I wanted to run the workflow. I know it is the problem with version of SAM-to-BAM. But, when I searched the Tool sheds > Search and browse tool sheds, there is only version 1.1.4 for SAM-to-BAM. When I tried to edit the workflow from Workflow > ...> Edit, I did not find where could I change the version of SAM-to-BAM. When I clicked on SAM-to-BAM on the Workflow Canvas, it showed the version is 1.1.3 for it. Could you tell me which version do I need exactly, and how to figure out this? Thanks a lot! Best, Xiaofei ___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
{kind=link}
participants (2)
-
 Nate Coraor
Nate Coraor -
 Wang, Xiaofei
Wang, Xiaofei