Why does my trackster complain about not being able to display BED files?

Hi, I am using the latest version of galaxy at my local installation and I try to use trackster to display a simple BED file, but it invariably complains about not having a converter for this datatype and it tells me to check my datatypes_conf.xml which I did (not sure what I should be looking for, but this is the section on BED files... Am I missing a converter? And why would a BED file even needed to be converted? Thanks Thon <datatype extension="bed" type="galaxy.datatypes.interval:Bed" display_in_upload="true"> <converter file="bed_to_gff_converter.xml" target_datatype="gff"/> <converter file="interval_to_coverage.xml" target_datatype="coverage"/> <converter file="bed_to_bgzip_converter.xml" target_datatype="bgzip"/> <converter file="bed_to_tabix_converter.xml" target_datatype="tabix" depends_on="bgzip"/> <converter file="bed_to_summary_tree_converter.xml" target_datatype="summary_tree"/> <converter file="bed_to_fli_converter.xml" target_datatype="fli"/> <!-- <display file="ucsc/interval_as_bed.xml" /> --> <!-- <display file="genetrack.xml" /> --> <display file="igb/bed.xml" /> </datatype> <datatype extension="bedgraph" type="galaxy.datatypes.interval:BedGraph" display_in_upload="true"> <converter file="bedgraph_to_bigwig_converter.xml" target_datatype="bigwig"/> </datatype> <datatype extension="bedstrict" type="galaxy.datatypes.interval:BedStrict" /> <!-- <datatype extension="bed6" type="galaxy.datatypes.interval:Bed6"> <converter file="bed_to_genetrack_converter.xml" target_datatype="genetrack"/> </datatype> --> <datatype extension="bed12" type="galaxy.datatypes.interval:Bed12" /> <datatype extension="len" type="galaxy.datatypes.chrominfo:ChromInfo" display_in_upload="true"> <converter file="len_to_linecount.xml" target_datatype="linecount" /> </datatype> <datatype extension="bigbed" type="galaxy.datatypes.binary:BigBed" mimetype="application/octet-stream" dis

Hi Anthonius, Do you have the BEDTools installed and its directory added to your PATH environment variable? Trackster uses compressed file formats to reduce the amount of data transfers. I hope this helps, Sam On Wed, Aug 21, 2013 at 4:11 PM, Anthonius deBoer <thondeboer@me.com> wrote:
Hi,
I am using the latest version of galaxy at my local installation and I try to use trackster to display a simple BED file, but it invariably complains about not having a converter for this datatype and it tells me to check my datatypes_conf.xml which I did (not sure what I should be looking for, but this is the section on BED files...
Am I missing a converter? And why would a BED file even needed to be converted?
Thanks
Thon
<datatype extension="bed" type="galaxy.datatypes.interval:Bed" display_in_upload="true"> <converter file="bed_to_gff_converter.xml" target_datatype="gff"/> <converter file="interval_to_coverage.xml" target_datatype="coverage"/> <converter file="bed_to_bgzip_converter.xml" target_datatype="bgzip"/> <converter file="bed_to_tabix_converter.xml" target_datatype="tabix" depends_on="bgzip"/> <converter file="bed_to_summary_tree_converter.xml" target_datatype="summary_tree"/> <converter file="bed_to_fli_converter.xml" target_datatype="fli"/> <!-- <display file="ucsc/interval_as_bed.xml" /> --> <!-- <display file="genetrack.xml" /> --> <display file="igb/bed.xml" /> </datatype> <datatype extension="bedgraph" type="galaxy.datatypes.interval:BedGraph" display_in_upload="true"> <converter file="bedgraph_to_bigwig_converter.xml" target_datatype="bigwig"/> </datatype> <datatype extension="bedstrict" type="galaxy.datatypes.interval:BedStrict" /> <!-- <datatype extension="bed6" type="galaxy.datatypes.interval:Bed6"> <converter file="bed_to_genetrack_converter.xml" target_datatype="genetrack"/> </datatype> --> <datatype extension="bed12" type="galaxy.datatypes.interval:Bed12" /> <datatype extension="len" type="galaxy.datatypes.chrominfo:ChromInfo" display_in_upload="true"> <converter file="len_to_linecount.xml" target_datatype="linecount" /> </datatype> <datatype extension="bigbed" type="galaxy.datatypes.binary:BigBed" mimetype="application/octet-stream" dis
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
Yep... $ bedtools --version bedtools v2.17.0 Thon On Aug 21, 2013, at 01:40 PM, sam guerler <aysam.guerler@gmail.com> wrote: Hi Anthonius, Do you have the BEDTools installed and its directory added to your PATH environment variable? Trackster uses compressed file formats to reduce the amount of data transfers. I hope this helps, Sam On Wed, Aug 21, 2013 at 4:11 PM, Anthonius deBoer <thondeboer@me.com> wrote: Hi, I am using the latest version of galaxy at my local installation and I try to use trackster to display a simple BED file, but it invariably complains about not having a converter for this datatype and it tells me to check my datatypes_conf.xml which I did (not sure what I should be looking for, but this is the section on BED files... Am I missing a converter? And why would a BED file even needed to be converted? Thanks Thon <datatype extension="bed" type="galaxy.datatypes.interval:Bed" display_in_upload="true"> <converter file="bed_to_gff_converter.xml" target_datatype="gff"/> <converter file="interval_to_coverage.xml" target_datatype="coverage"/> <converter file="bed_to_bgzip_converter.xml" target_datatype="bgzip"/> <converter file="bed_to_tabix_converter.xml" target_datatype="tabix" depends_on="bgzip"/> <converter file="bed_to_summary_tree_converter.xml" target_datatype="summary_tree"/> <converter file="bed_to_fli_converter.xml" target_datatype="fli"/> <!-- <display file="ucsc/interval_as_bed.xml" /> --> <!-- <display file="genetrack.xml" /> --> <display file="igb/bed.xml" /> </datatype> <datatype extension="bedgraph" type="galaxy.datatypes.interval:BedGraph" display_in_upload="true"> <converter file="bedgraph_to_bigwig_converter.xml" target_datatype="bigwig"/> </datatype> <datatype extension="bedstrict" type="galaxy.datatypes.interval:BedStrict" /> <!-- <datatype extension="bed6" type="galaxy.datatypes.interval:Bed6"> <converter file="bed_to_genetrack_converter.xml" target_datatype="genetrack"/> </datatype> --> <datatype extension="bed12" type="galaxy.datatypes.interval:Bed12" /> <datatype extension="len" type="galaxy.datatypes.chrominfo:ChromInfo" display_in_upload="true"> <converter file="len_to_linecount.xml" target_datatype="linecount" /> </datatype> <datatype extension="bigbed" type="galaxy.datatypes.binary:BigBed" mimetype="application/octet-stream" dis ___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/

Hi Thon double check your datatypes_conf.xml (which was not touched/changed during the update process, since it is listed in the .hgignore file, I guess) with the new datatypes_conf.xml.sample file for any changes regarding converters for "bed" files Regards, Hans-Rudolf On 08/22/2013 02:51 AM, Anthonius deBoer wrote:
Yep...
$ bedtools --version bedtools v2.17.0
Thon
On Aug 21, 2013, at 01:40 PM, sam guerler <aysam.guerler@gmail.com> wrote:
Hi Anthonius,
Do you have the BEDTools installed and its directory added to your PATH environment variable? Trackster uses compressed file formats to reduce the amount of data transfers.
I hope this helps, Sam
On Wed, Aug 21, 2013 at 4:11 PM, Anthonius deBoer <thondeboer@me.com <mailto:thondeboer@me.com>> wrote:
Hi,
I am using the latest version of galaxy at my local installation and I try to use trackster to display a simple BED file, but it invariably complains about not having a converter for this datatype and it tells me to check my datatypes_conf.xml which I did (not sure what I should be looking for, but this is the section on BED files...
Am I missing a converter? And why would a BED file even needed to be converted?
Thanks
Thon
<datatype extension="bed" type="galaxy.datatypes.interval:Bed" display_in_upload="true"> <converter file="bed_to_gff_converter.xml" target_datatype="gff"/> <converter file="interval_to_coverage.xml" target_datatype="coverage"/> <converter file="bed_to_bgzip_converter.xml" target_datatype="bgzip"/> <converter file="bed_to_tabix_converter.xml" target_datatype="tabix" depends_on="bgzip"/> <converter file="bed_to_summary_tree_converter.xml" target_datatype="summary_tree"/> <converter file="bed_to_fli_converter.xml" target_datatype="fli"/> <!-- <display file="ucsc/interval_as_bed.xml" /> --> <!-- <display file="genetrack.xml" /> --> <display file="igb/bed.xml" /> </datatype> <datatype extension="bedgraph" type="galaxy.datatypes.interval:BedGraph" display_in_upload="true"> <converter file="bedgraph_to_bigwig_converter.xml" target_datatype="bigwig"/> </datatype> <datatype extension="bedstrict" type="galaxy.datatypes.interval:BedStrict" /> <!-- <datatype extension="bed6" type="galaxy.datatypes.interval:Bed6"> <converter file="bed_to_genetrack_converter.xml" target_datatype="genetrack"/> </datatype> --> <datatype extension="bed12" type="galaxy.datatypes.interval:Bed12" /> <datatype extension="len" type="galaxy.datatypes.chrominfo:ChromInfo" display_in_upload="true"> <converter file="len_to_linecount.xml" target_datatype="linecount" /> </datatype> <datatype extension="bigbed" type="galaxy.datatypes.binary:BigBed" mimetype="application/octet-stream" dis
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/
___________________________________________________________ Please keep all replies on the list by using "reply all" in your mail client. To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
To search Galaxy mailing lists use the unified search at: http://galaxyproject.org/search/mailinglists/

Am I missing a converter?
Yes, you are missing the bed_to_bigwig converter. If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters. If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters.
And why would a BED file even needed to be converted?
Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast. J.
Thanks Jeremy, I did what you suggested and now it no longer complains! Not sure if I had edited datatypes_conf.xml but just copying the sample worked and let's just hope I did not break any of my changes Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: Jeremy Goecks [mailto:jeremy.goecks@emory.edu] Sent: Thursday, August 22, 2013 11:11 AM To: Anthonius deBoer Cc: sam guerler; Galaxy-dev Galaxy-dev Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Am I missing a converter? Yes, you are missing the bed_to_bigwig converter. If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters. If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters. And why would a BED file even needed to be converted? Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast. J.
I now have a different problem with a different (production) version of galaxy trickster. I always get this message "Could not load chroms for this dbkey" I have checked that I have files with the [key].len in tool-data/shared/ucsc/chrom and I have twobit files etc. The weird thing is, that the "Create visualization" shows the genome keys I have but this window shows nothing for the dbkey it things it should be getting. So somewhere it "looses" the key. Any ideas? (I'm running the latest update) Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 11:02 AM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Thanks Jeremy, I did what you suggested and now it no longer complains! Not sure if I had edited datatypes_conf.xml but just copying the sample worked and let's just hope I did not break any of my changes Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: Jeremy Goecks [mailto:jeremy.goecks@emory.edu] Sent: Thursday, August 22, 2013 11:11 AM To: Anthonius deBoer Cc: sam guerler; Galaxy-dev Galaxy-dev Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Am I missing a converter? Yes, you are missing the bed_to_bigwig converter. If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters. If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters. And why would a BED file even needed to be converted? Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast. J.
{kind=link}
I tracked it down to a problem with the API proxy configuration. I had the API calls diverted to a different server since I wanted to ensure that API calls would be handled by a different server, but that does not seem to work correctly for the API calls used in trickster. Issue resolved. Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:12 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? I now have a different problem with a different (production) version of galaxy trickster. I always get this message "Could not load chroms for this dbkey" I have checked that I have files with the [key].len in tool-data/shared/ucsc/chrom and I have twobit files etc. The weird thing is, that the "Create visualization" shows the genome keys I have but this window shows nothing for the dbkey it things it should be getting. So somewhere it "looses" the key. Any ideas? (I'm running the latest update) Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: galaxy-dev-bounces@lists.bx.psu.edu <mailto:galaxy-dev-bounces@lists.bx.psu.edu> [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 11:02 AM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Thanks Jeremy, I did what you suggested and now it no longer complains! Not sure if I had edited datatypes_conf.xml but just copying the sample worked and let's just hope I did not break any of my changes Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: Jeremy Goecks [mailto:jeremy.goecks@emory.edu] Sent: Thursday, August 22, 2013 11:11 AM To: Anthonius deBoer Cc: sam guerler; Galaxy-dev Galaxy-dev Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Am I missing a converter? Yes, you are missing the bed_to_bigwig converter. If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters. If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters. And why would a BED file even needed to be converted? Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast. J.
{kind=link}
Ofcourse I spoke too soon. I get a problem in that the conversion of my dataset reports that it cannot find the chromosome length files, even though trickster itself has no problem showing it. I looked at the problem script and it is showing below It seems that the script is passed the path to the chromosome files like this "tool-data/shared/ucsc/chrom/hg19-decoy.len" This will clearly never work since this file is not relative to the working directory.. SO somewhere a script forgets to add the ${GALAXY_DATA_DIR} or whatever that parameter is... I could probably hardcode the location of the tool-data directory, but I don't think that should be the solution. Why do I only have this issue? SHOULD I have hardcoded the location of tool-data? The universe_wgi.ini.sample file does not hardcode this. Am I missing some updates? Thanks, Thon #!/bin/sh GALAXY_LIB="None" if [ "$GALAXY_LIB" != "None" ]; then if [ -n "$PYTHONPATH" ]; then PYTHONPATH="$GALAXY_LIB:$PYTHONPATH" else PYTHONPATH="$GALAXY_LIB" fi export PYTHONPATH fi TMPDIR=/mnt/ngs/analysis/svcgalaxy/DATATEST/TMP export TMPDIR cd /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 grep -v '^#' /mnt/ngs/analysis/svcgalaxy/DATATEST/files/002/dataset_2288.dat | sort -k1,1 | bedtools genomecov -bg -split -i stdin -g tool-data/shared/ucsc/chrom/hg19-decoy.len > temp.bg ; bedGraphToBigWig temp.bg tool-data/shared/ucsc/chrom/hg19-decoy.len /mnt/ngs/analysis/svcgalaxy/DATATEST/files/002/dataset_2315.dat; cd /mnt/ngs/analysis/svcgalaxy/galaxy-test; /mnt/ngs/analysis/svcgalaxy/galaxy-test/set_metadata.sh /mnt/ngs/analysis/svcgalaxy/DATATEST/files /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 . /mnt/ngs/analysis/svcgalaxy/galaxy-test/universe_wsgi.ini /mnt/ngs/analysis/svcgalaxy/DATA/TMP/tmp2KAn7W /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy.j son /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata _in_HistoryDatasetAssociation_2551_XsCZer,/mnt/ngs/analysis/svcgalaxy/DATATE ST/job_working_directory/001/1916/metadata_kwds_HistoryDatasetAssociation_25 51_iNUAr_,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/191 6/metadata_out_HistoryDatasetAssociation_2551_ZHhosg,/mnt/ngs/analysis/svcga laxy/DATATEST/job_working_directory/001/1916/metadata_results_HistoryDataset Association_2551_tqtiIX,,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_di rectory/001/1916/metadata_override_HistoryDatasetAssociation_2551_MOfd8w echo $? > /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy_1 916.ec Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:47 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? I tracked it down to a problem with the API proxy configuration. I had the API calls diverted to a different server since I wanted to ensure that API calls would be handled by a different server, but that does not seem to work correctly for the API calls used in trickster. Issue resolved. Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: galaxy-dev-bounces@lists.bx.psu.edu <mailto:galaxy-dev-bounces@lists.bx.psu.edu> [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:12 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? I now have a different problem with a different (production) version of galaxy trickster. I always get this message "Could not load chroms for this dbkey" I have checked that I have files with the [key].len in tool-data/shared/ucsc/chrom and I have twobit files etc. The weird thing is, that the "Create visualization" shows the genome keys I have but this window shows nothing for the dbkey it things it should be getting. So somewhere it "looses" the key. Any ideas? (I'm running the latest update) Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: galaxy-dev-bounces@lists.bx.psu.edu <mailto:galaxy-dev-bounces@lists.bx.psu.edu> [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 11:02 AM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Thanks Jeremy, I did what you suggested and now it no longer complains! Not sure if I had edited datatypes_conf.xml but just copying the sample worked and let's just hope I did not break any of my changes Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: <mailto:thondeboer@me.com> thondeboer@me.com Public profile on <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> LinkedIn From: Jeremy Goecks [mailto:jeremy.goecks@emory.edu] Sent: Thursday, August 22, 2013 11:11 AM To: Anthonius deBoer Cc: sam guerler; Galaxy-dev Galaxy-dev Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Am I missing a converter? Yes, you are missing the bed_to_bigwig converter. If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters. If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters. And why would a BED file even needed to be converted? Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast. J.
{kind=link}
It appears you are missing some updates. This issue was fixed in this changeset 9992: -- changeset: 9992:7105c53139d4 branch: stable parent: 9990:f35c0441e374 user: jeremy goecks <jeremy.goecks@emory.edu> date: Tue Jun 11 17:34:26 2013 -0400 files: lib/galaxy/tools/actions/__init__.py description: Use len_file_path config option rather than hardcoded path for chromInfo tool parameter. -- You might have missed this changeset if you updated to the late June release soon after it came out. However, it is definitely included in the August release. Can you confirm that you're running the August release? Thanks, J. On Aug 23, 2013, at 9:08 PM, Thon de Boer wrote:
Ofcourse I spoke too soon…
I get a problem in that the conversion of my dataset reports that it cannot find the chromosome length files, even though trickster itself has no problem showing it…
I looked at the problem script and it is showing below
It seems that the script is passed the path to the chromosome files like this “tool-data/shared/ucsc/chrom/hg19-decoy.len”
This will clearly never work since this file is not relative to the working directory.. SO somewhere a script forgets to add the ${GALAXY_DATA_DIR} or whatever that parameter is…..
I could probably hardcode the location of the tool-data directory, but I don’t think that should be the solution…
Why do I only have this issue? SHOULD I have hardcoded the location of tool-data?
The universe_wgi.ini.sample file does not hardcode this…
Am I missing some updates?
Thanks,
Thon
#!/bin/sh GALAXY_LIB="None" if [ "$GALAXY_LIB" != "None" ]; then if [ -n "$PYTHONPATH" ]; then PYTHONPATH="$GALAXY_LIB:$PYTHONPATH" else PYTHONPATH="$GALAXY_LIB" fi export PYTHONPATH fi TMPDIR=/mnt/ngs/analysis/svcgalaxy/DATATEST/TMP export TMPDIR
cd /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 grep -v '^#' /mnt/ngs/analysis/svcgalaxy/DATATEST/files/002/dataset_2288.dat | sort -k1,1 | bedtools genomecov -bg -split -i stdin -gtool-data/shared/ucsc/chrom/hg19-decoy.len > temp.bg ; bedGraphToBigWig temp.bg tool-data/shared/ucsc/chrom/hg19-decoy.len/mnt/ngs/analysis/svcgalaxy/DATATEST/files/002/dataset_2315.dat; cd /mnt/ngs/analysis/svcgalaxy/galaxy-test; /mnt/ngs/analysis/svcgalaxy/galaxy-test/set_metadata.sh /mnt/ngs/analysis/svcgalaxy/DATATEST/files /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 . /mnt/ngs/analysis/svcgalaxy/galaxy-test/universe_wsgi.ini /mnt/ngs/analysis/svcgalaxy/DATA/TMP/tmp2KAn7W /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy.json /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata_in_HistoryDatasetAssociation_2551_XsCZer,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata_kwds_HistoryDatasetAssociation_2551_iNUAr_,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata_out_HistoryDatasetAssociation_2551_ZHhosg,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata_results_HistoryDatasetAssociation_2551_tqtiIX,,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata_override_HistoryDatasetAssociation_2551_MOfd8w echo $? > /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy_1916.ec
Regards,
Thon
Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com Public profile on LinkedIn
From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:47 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files?
I tracked it down to a problem with the API proxy configuration.
I had the API calls diverted to a different server since I wanted to ensure that API calls would be handled by a different server, but that does not seem to work correctly for the API calls used in trickster…
Issue resolved…
Regards,
Thon
Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com Public profile on LinkedIn
From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:12 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files?
I now have a different problem with a different (production) version of galaxy trickster.
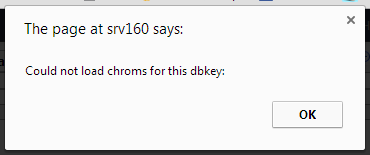
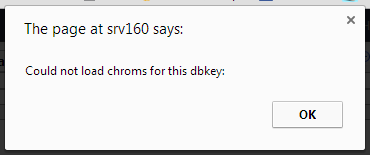
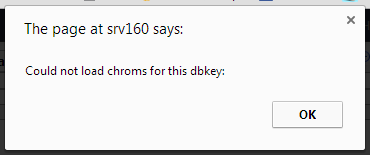
I always get this message “Could not load chroms for this dbkey”
I have checked that I have files with the [key].len in tool-data/shared/ucsc/chrom and I have twobit files etc.
The weird thing is, that the “Create visualization” shows the genome keys I have but this window shows nothing for the dbkey it things it should be getting…
So somewhere it “looses” the key…
Any ideas?
(I’m running the latest update)
<image001.png>
Regards,
Thon
Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com Public profile on LinkedIn
From: galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 11:02 AM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files?
Thanks Jeremy,
I did what you suggested and now it no longer complains! Not sure if I had edited datatypes_conf.xml but just copying the sample worked and let’s just hope I did not break any of my changes
Regards,
Thon
Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com Public profile on LinkedIn
From: Jeremy Goecks [mailto:jeremy.goecks@emory.edu] Sent: Thursday, August 22, 2013 11:11 AM To: Anthonius deBoer Cc: sam guerler; Galaxy-dev Galaxy-dev Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files?
Am I missing a converter?
Yes, you are missing the bed_to_bigwig converter.
If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters.
If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters.
And why would a BED file even needed to be converted?
Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast.
J.
Yeah, pretty sure I'm up to date. $ hg branches default 10411:c42567f43aa7 stable 10408:6822f41bc9bb (inactive) [svcbioinfo@srv160 gs]$ hg branch stable [svcbioinfo@srv160 gs]$ hg tip changeset: 10411:c42567f43aa7 tag: tip user: greg date: Mon Aug 19 13:19:56 2013 -0400 summary: Filter invalid objects when generating the list of repository_dependencies objects that are associated with a tool shed repository installed into Galaxy. [svcbioinfo@srv160 gs]$ hg incoming warning: bitbucket.org certificate with fingerprint 24:9c:45:8b:9c:aa:ba:55:4e:01:6d:58:ff:e4:28:7d:2a:14:ae:3b not verified (check hostfingerprints or web.cacerts config setting) comparing with https://bitbucket.org/galaxy/galaxy-dist/ searching for changes no changes found $ hg update release_2013.08.12 0 files updated, 0 files merged, 0 files removed, 0 files unresolved Should I just take "lib/galaxy/tools/actions/__init__.py " from a fresh install? Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: Jeremy Goecks [mailto:jeremy.goecks@emory.edu] Sent: Saturday, August 24, 2013 7:39 AM To: Thon de Boer Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? It appears you are missing some updates. This issue was fixed in this changeset 9992: -- changeset: 9992:7105c53139d4 branch: stable parent: 9990:f35c0441e374 user: jeremy goecks <jeremy.goecks@emory.edu <mailto:jeremy.goecks@emory.edu> > date: Tue Jun 11 17:34:26 2013 -0400 files: lib/galaxy/tools/actions/__init__.py description: Use len_file_path config option rather than hardcoded path for chromInfo tool parameter. -- You might have missed this changeset if you updated to the late June release soon after it came out. However, it is definitely included in the August release. Can you confirm that you're running the August release? Thanks, J. On Aug 23, 2013, at 9:08 PM, Thon de Boer wrote: Ofcourse I spoke too soon. I get a problem in that the conversion of my dataset reports that it cannot find the chromosome length files, even though trickster itself has no problem showing it. I looked at the problem script and it is showing below It seems that the script is passed the path to the chromosome files like this "tool-data/shared/ucsc/chrom/hg19-decoy.len" This will clearly never work since this file is not relative to the working directory.. SO somewhere a script forgets to add the ${GALAXY_DATA_DIR} or whatever that parameter is... I could probably hardcode the location of the tool-data directory, but I don't think that should be the solution. Why do I only have this issue? SHOULD I have hardcoded the location of tool-data? The universe_wgi.ini.sample file does not hardcode this. Am I missing some updates? Thanks, Thon #!/bin/sh GALAXY_LIB="None" if [ "$GALAXY_LIB" != "None" ]; then if [ -n "$PYTHONPATH" ]; then PYTHONPATH="$GALAXY_LIB:$PYTHONPATH" else PYTHONPATH="$GALAXY_LIB" fi export PYTHONPATH fi TMPDIR=/mnt/ngs/analysis/svcgalaxy/DATATEST/TMP export TMPDIR cd /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 grep -v '^#' /mnt/ngs/analysis/svcgalaxy/DATATEST/files/002/dataset_2288.dat | sort -k1,1 | bedtools genomecov -bg -split -i stdin -gtool-data/shared/ucsc/chrom/hg19-decoy.len > temp.bg ; bedGraphToBigWig temp.bg tool-data/shared/ucsc/chrom/hg19-decoy.len/mnt/ngs/analysis/svcgalaxy/DATATE ST/files/002/dataset_2315.dat; cd /mnt/ngs/analysis/svcgalaxy/galaxy-test; /mnt/ngs/analysis/svcgalaxy/galaxy-test/set_metadata.sh /mnt/ngs/analysis/svcgalaxy/DATATEST/files /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 . /mnt/ngs/analysis/svcgalaxy/galaxy-test/universe_wsgi.ini /mnt/ngs/analysis/svcgalaxy/DATA/TMP/tmp2KAn7W /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy.j son /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata _in_HistoryDatasetAssociation_2551_XsCZer,/mnt/ngs/analysis/svcgalaxy/DATATE ST/job_working_directory/001/1916/metadata_kwds_HistoryDatasetAssociation_25 51_iNUAr_,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/191 6/metadata_out_HistoryDatasetAssociation_2551_ZHhosg,/mnt/ngs/analysis/svcga laxy/DATATEST/job_working_directory/001/1916/metadata_results_HistoryDataset Association_2551_tqtiIX,,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_di rectory/001/1916/metadata_override_HistoryDatasetAssociation_2551_MOfd8w echo $? > /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy_1 916.ec Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: <mailto:galaxy-dev-bounces@lists.bx.psu.edu> galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:47 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? I tracked it down to a problem with the API proxy configuration. I had the API calls diverted to a different server since I wanted to ensure that API calls would be handled by a different server, but that does not seem to work correctly for the API calls used in trickster. Issue resolved. Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: <mailto:galaxy-dev-bounces@lists.bx.psu.edu> galaxy-dev-bounces@lists.bx.psu.edu [ <mailto:galaxy-dev-bounces@lists.bx.psu.edu> mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:12 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? I now have a different problem with a different (production) version of galaxy trickster. I always get this message "Could not load chroms for this dbkey" I have checked that I have files with the [key].len in tool-data/shared/ucsc/chrom and I have twobit files etc. The weird thing is, that the "Create visualization" shows the genome keys I have but this window shows nothing for the dbkey it things it should be getting. So somewhere it "looses" the key. Any ideas? (I'm running the latest update) <image001.png> Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: <mailto:galaxy-dev-bounces@lists.bx.psu.edu> galaxy-dev-bounces@lists.bx.psu.edu [ <mailto:galaxy-dev-bounces@lists.bx.psu.edu> mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 11:02 AM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Thanks Jeremy, I did what you suggested and now it no longer complains! Not sure if I had edited datatypes_conf.xml but just copying the sample worked and let's just hope I did not break any of my changes Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: Jeremy Goecks [ <mailto:jeremy.goecks@emory.edu> mailto:jeremy.goecks@emory.edu] Sent: Thursday, August 22, 2013 11:11 AM To: Anthonius deBoer Cc: sam guerler; Galaxy-dev Galaxy-dev Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Am I missing a converter? Yes, you are missing the bed_to_bigwig converter. If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters. If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters. And why would a BED file even needed to be converted? Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast. J.
Yeah.I definitely have an issue with the update. It seems my __init___.py has not been updated. Wonder what else is not updated.. Seems there are more changes I don't have if I do diff galaxy-CLEAN/lib/galaxy/tools/actions/ galaxy-colo/lib/galaxy/tools/actions/ But these are the differences with __init.py__ Sigh.Guess I have to re-install from scratch and make all my changes again. Thon diff galaxy-CLEAN/lib/galaxy/tools/actions/__init__.py galaxy-colo/lib/galaxy/tools/actions/__init__.py 1,4d0 < import os < import galaxy.tools < < from galaxy.exceptions import ObjectInvalid 6,10c2 < from galaxy.tools.parameters import DataToolParameter, SelectToolParameter < from galaxy.tools.parameters.grouping import Conditional, Repeat < from galaxy.util.json import from_json_string < from galaxy.util.json import to_json_string < from galaxy.util.none_like import NoneDataset ---
from galaxy.util.bunch import Bunch
11a4,6
from galaxy.util.json import to_json_string
from galaxy.tools.parameters import *
from galaxy.tools.parameters.grouping import *
12a8
from galaxy.util.none_like import NoneDataset
13a10,12
from galaxy.exceptions import ObjectInvalid
import galaxy.tools
from types import *
38,39d36 < if not data: < return data 42c39 < if not data.datatype.matches_any( formats ): ---
if data and not isinstance( data.datatype, formats ):
60c57 < if not trans.app.security_agent.can_access_dataset( current_user_roles, data.dataset ): ---
if data and not
trans.app.security_agent.can_access_dataset( current_user_roles, data.dataset ): 76c73 < if not new_data or new_data.datatype.matches_any( conversion_datatypes ): ---
if not new_data or isinstance(
new_data.datatype, conversion_datatypes ): 96c93 < if not new_data or new_data.datatype.matches_any( conversion_datatypes ): ---
if not new_data or isinstance( new_data.datatype,
conversion_datatypes ): 111c108 < def execute(self, tool, trans, incoming={}, return_job=False, set_output_hid=True, set_output_history=True, history=None, job_params=None, rerun_remap_job_id=None): ---
def execute(self, tool, trans, incoming={}, return_job=False,
set_output_hid=True, set_output_history=True, history=None, job_params=None ): 220c217 < chrom_info = os.path.join( trans.app.config.len_file_path, "%s.len" % input_dbkey ) ---
chrom_info = os.path.join(
trans.app.config.tool_data_path, 'shared','ucsc','chrom', "%s.len" % input_dbkey ) 277,279c274 < input_dataset = inp_data[output.format_source] < input_extension = input_dataset.ext < ext = input_extension ---
ext = inp_data[output.format_source].ext
413,446d407 < # Now that we have a job id, we can remap any outputs if this is a rerun and the user chose to continue dependent jobs < # This functionality requires tracking jobs in the database. < if trans.app.config.track_jobs_in_database and rerun_remap_job_id is not None: < try: < old_job = trans.sa_session.query( trans.app.model.Job ).get(rerun_remap_job_id) < assert old_job is not None, '(%s/%s): Old job id is invalid' % (rerun_remap_job_id, job.id) < assert old_job.tool_id == job.tool_id, '(%s/%s): Old tool id (%s) does not match rerun tool id (%s)' % (old_job.id, job.id, old_job.tool_id, job.tool_id) < if trans.user is not None: < assert old_job.user_id == trans.user.id, '(%s/%s): Old user id (%s) does not match rerun user id (%s)' % (old_job.id, job.id, old_job.user_id, trans.user.id) < elif trans.user is None and type( galaxy_session ) == trans.model.GalaxySession: < assert old_job.session_id == galaxy_session.id, '(%s/%s): Old session id (%s) does not match rerun session id (%s)' % (old_job.id, job.id, old_job.session_id, galaxy_session.id) < else: < raise Exception('(%s/%s): Remapping via the API is not (yet) supported' % (old_job.id, job.id)) < for jtod in old_job.output_datasets: < for (job_to_remap, jtid) in [(jtid.job, jtid) for jtid in jtod.dataset.dependent_jobs]: < if (trans.user is not None and job_to_remap.user_id == trans.user.id) or (trans.user is None and job_to_remap.session_id == galaxy_session.id): < if job_to_remap.state == job_to_remap.states.PAUSED: < job_to_remap.state = job_to_remap.states.NEW < for hda in [ dep_jtod.dataset for dep_jtod in job_to_remap.output_datasets ]: < if hda.state == hda.states.PAUSED: < hda.state = hda.states.NEW < hda.info = None < for p in job_to_remap.parameters: < if p.name == jtid.name and p.value == str(jtod.dataset.id): < p.value = str(out_data[jtod.name].id) < jtid.dataset = out_data[jtod.name] < jtid.dataset.hid = jtod.dataset.hid < log.info('Job %s input HDA %s remapped to new HDA %s' % (job_to_remap.id, jtod.dataset.id, jtid.dataset.id)) < trans.sa_session.add(job_to_remap) < trans.sa_session.add(jtid) < jtod.dataset.visible = False < trans.sa_session.add(jtod) < except Exception, e: < log.exception('Cannot remap rerun dependencies.') Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: Jeremy Goecks [mailto:jeremy.goecks@emory.edu] Sent: Saturday, August 24, 2013 7:39 AM To: Thon de Boer Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? It appears you are missing some updates. This issue was fixed in this changeset 9992: -- changeset: 9992:7105c53139d4 branch: stable parent: 9990:f35c0441e374 user: jeremy goecks <jeremy.goecks@emory.edu <mailto:jeremy.goecks@emory.edu> > date: Tue Jun 11 17:34:26 2013 -0400 files: lib/galaxy/tools/actions/__init__.py description: Use len_file_path config option rather than hardcoded path for chromInfo tool parameter. -- You might have missed this changeset if you updated to the late June release soon after it came out. However, it is definitely included in the August release. Can you confirm that you're running the August release? Thanks, J. On Aug 23, 2013, at 9:08 PM, Thon de Boer wrote: Ofcourse I spoke too soon. I get a problem in that the conversion of my dataset reports that it cannot find the chromosome length files, even though trickster itself has no problem showing it. I looked at the problem script and it is showing below It seems that the script is passed the path to the chromosome files like this "tool-data/shared/ucsc/chrom/hg19-decoy.len" This will clearly never work since this file is not relative to the working directory.. SO somewhere a script forgets to add the ${GALAXY_DATA_DIR} or whatever that parameter is... I could probably hardcode the location of the tool-data directory, but I don't think that should be the solution. Why do I only have this issue? SHOULD I have hardcoded the location of tool-data? The universe_wgi.ini.sample file does not hardcode this. Am I missing some updates? Thanks, Thon #!/bin/sh GALAXY_LIB="None" if [ "$GALAXY_LIB" != "None" ]; then if [ -n "$PYTHONPATH" ]; then PYTHONPATH="$GALAXY_LIB:$PYTHONPATH" else PYTHONPATH="$GALAXY_LIB" fi export PYTHONPATH fi TMPDIR=/mnt/ngs/analysis/svcgalaxy/DATATEST/TMP export TMPDIR cd /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 grep -v '^#' /mnt/ngs/analysis/svcgalaxy/DATATEST/files/002/dataset_2288.dat | sort -k1,1 | bedtools genomecov -bg -split -i stdin -gtool-data/shared/ucsc/chrom/hg19-decoy.len > temp.bg ; bedGraphToBigWig temp.bg tool-data/shared/ucsc/chrom/hg19-decoy.len/mnt/ngs/analysis/svcgalaxy/DATATE ST/files/002/dataset_2315.dat; cd /mnt/ngs/analysis/svcgalaxy/galaxy-test; /mnt/ngs/analysis/svcgalaxy/galaxy-test/set_metadata.sh /mnt/ngs/analysis/svcgalaxy/DATATEST/files /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916 . /mnt/ngs/analysis/svcgalaxy/galaxy-test/universe_wsgi.ini /mnt/ngs/analysis/svcgalaxy/DATA/TMP/tmp2KAn7W /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy.j son /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/metadata _in_HistoryDatasetAssociation_2551_XsCZer,/mnt/ngs/analysis/svcgalaxy/DATATE ST/job_working_directory/001/1916/metadata_kwds_HistoryDatasetAssociation_25 51_iNUAr_,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/191 6/metadata_out_HistoryDatasetAssociation_2551_ZHhosg,/mnt/ngs/analysis/svcga laxy/DATATEST/job_working_directory/001/1916/metadata_results_HistoryDataset Association_2551_tqtiIX,,/mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_di rectory/001/1916/metadata_override_HistoryDatasetAssociation_2551_MOfd8w echo $? > /mnt/ngs/analysis/svcgalaxy/DATATEST/job_working_directory/001/1916/galaxy_1 916.ec Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: <mailto:galaxy-dev-bounces@lists.bx.psu.edu> galaxy-dev-bounces@lists.bx.psu.edu [mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:47 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? I tracked it down to a problem with the API proxy configuration. I had the API calls diverted to a different server since I wanted to ensure that API calls would be handled by a different server, but that does not seem to work correctly for the API calls used in trickster. Issue resolved. Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: <mailto:galaxy-dev-bounces@lists.bx.psu.edu> galaxy-dev-bounces@lists.bx.psu.edu [ <mailto:galaxy-dev-bounces@lists.bx.psu.edu> mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 5:12 PM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? I now have a different problem with a different (production) version of galaxy trickster. I always get this message "Could not load chroms for this dbkey" I have checked that I have files with the [key].len in tool-data/shared/ucsc/chrom and I have twobit files etc. The weird thing is, that the "Create visualization" shows the genome keys I have but this window shows nothing for the dbkey it things it should be getting. So somewhere it "looses" the key. Any ideas? (I'm running the latest update) <image001.png> Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: <mailto:galaxy-dev-bounces@lists.bx.psu.edu> galaxy-dev-bounces@lists.bx.psu.edu [ <mailto:galaxy-dev-bounces@lists.bx.psu.edu> mailto:galaxy-dev-bounces@lists.bx.psu.edu] On Behalf Of Thon de Boer Sent: Friday, August 23, 2013 11:02 AM To: 'Jeremy Goecks' Cc: 'Galaxy-dev Galaxy-dev' Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Thanks Jeremy, I did what you suggested and now it no longer complains! Not sure if I had edited datatypes_conf.xml but just copying the sample worked and let's just hope I did not break any of my changes Regards, Thon Thon deBoer Ph.D., Bioinformatics Guru California, USA |p: +1 (650) 799-6839 |m: thondeboer@me.com <mailto:thondeboer@me.com> Public profile on LinkedIn <http://www.linkedin.com/pub/thon-de-boer/1/1ba/a5b/> From: Jeremy Goecks [ <mailto:jeremy.goecks@emory.edu> mailto:jeremy.goecks@emory.edu] Sent: Thursday, August 22, 2013 11:11 AM To: Anthonius deBoer Cc: sam guerler; Galaxy-dev Galaxy-dev Subject: Re: [galaxy-dev] Why does my trackster complain about not being able to display BED files? Am I missing a converter? Yes, you are missing the bed_to_bigwig converter. If you haven't made changes to your datatypes_conf.xml file, you can just copy datatypes_conf.xml.sample to datatypes_conf.xml to get the needed converters. If you've made changes to datatypes_conf.xml, you'll need to manually add the needed converters. We recently transitioned all the *_to_summary_tree converters to *_to_bigwig, so you'll want to remove the summary_tree converters and replace them with the bigwig converters. And why would a BED file even needed to be converted? Converter = indexer for visualizations. Datasets are indexed so that (a) aggregate (coverage) data is readily available when viewing a large region and (b) finding features in a small region (when zoomed in) is fast. J.
participants (5)
-
 Anthonius deBoer
Anthonius deBoer -
 Hans-Rudolf Hotz
Hans-Rudolf Hotz -
 Jeremy Goecks
Jeremy Goecks -
 sam guerler
sam guerler -
Thon de Boer