
Vasu, It's not clear what you're trying to accomplish, but I'll try to provide some information that you might find useful:
I want to include the following discussion in my message regarding use Bam files of Tophat to visualize reads either in IGV or Galaxy or other tools. I want to find out if I can plot RPKM/FPKM normalized values after running differential analysis in Cufflinks.
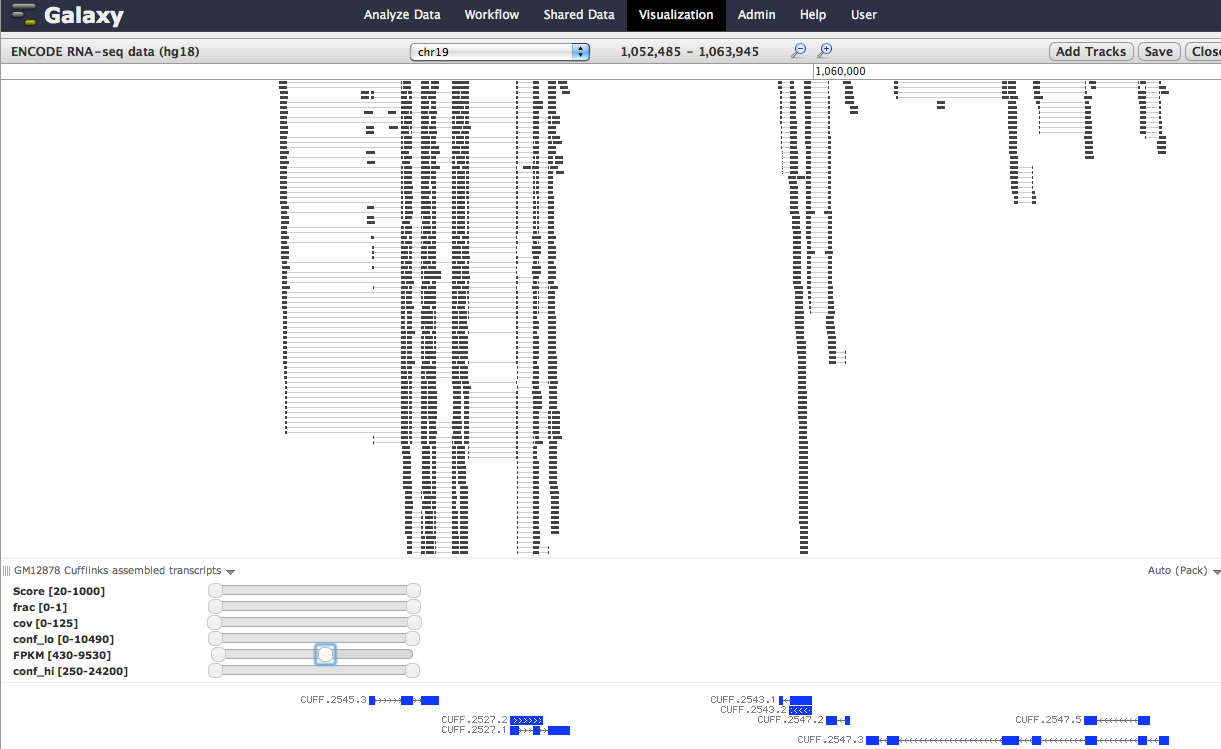
Galaxy has a number of tools for analyzing numerical data; look under the menu items Statistics and Graph/Display Data for useful tools. If you're looking to plot FPKM values in addition to mapped reads from Tophat and Cufflinks transcripts, the Galaxy Tracks Browser might prove useful as it has filtering functionality so that you can move a slider to show/hide data based on FPKM values; its often useful to use the sliders for FPKM measures to get a sense of your data. See the attached screenshot for an example.
On the seqanswer (http://seqanswers.com/forums/showthread.php?t=9947 ) there is a preliminary discussion about this how we can plot RPKM values to show the differential abundance in samples. Do we have any of such functionality in Galaxy? alternatively, I would like to have suggestions on the topic especially to normalize per million reads.
This thread discusses alternative approaches for computing RPKM/FPKM from mapped read data. If you choose not to use Cufflinks/compare/diff to compute RPKM, you can compute a rough RPKM by doing the following: (1) use the pileup tool to compute coverage across a genome; (2) divide coverage by the total number of mapped reads (in millions) to get coverage per million reads (CPM); and (3) average CPM across a transcript and divide by the length of the transcript to get ~RPKM. Best, J.
{kind=link}