
Hi, I am trying to analyze some RNA-seq data using the Galaxy. The reads for each sample comes from 3 lanes, so I need to integrate these three lane reads to be able to make a comparison between my samples. How do I 'put together' these three read-files? Is there any tools available in Galaxy for that, which I have missed? Unfortunately I do not have any programming skills. Many thanks, Mehdi M ---------------------------------------- Mehdi Motallebipour, PhD Mammalian Neurogenesis MRC Clinical Sciences Centre Imperial College London Hammersmith Hospital Campus Du Cane Road London W12 0NN United Kingdom Tel: +44-20-8383 8285

Hello Medhi, Sorry for the slow reply. It's not clear from your question, but let's assume that you have 3 lanes of RNA-seq data for each of your samples. In this case, here's a reasonable approach for analyzing this data: (1) Run Tophat + Cufflinks on each lane of data; (2) Use Cuffcompare on *all* transcripts produced by Cufflinks for all data; (3) Run Cuffdiff using (a) the combined transcripts produced by Cuffcompare and (b) segmenting the mapped reads produced by Tophat into groups based on the sample that they came from. Let us know if you have more questions. Thanks, J. On Feb 1, 2011, at 8:54 AM, Motallebipour, Mehdi wrote:
Hi,
I am trying to analyze some RNA-seq data using the Galaxy. The reads for each sample comes from 3 lanes, so I need to integrate these three lane reads to be able to make a comparison between my samples. How do I 'put together' these three read-files? Is there any tools available in Galaxy for that, which I have missed? Unfortunately I do not have any programming skills.
Many thanks, Mehdi M ---------------------------------------- Mehdi Motallebipour, PhD Mammalian Neurogenesis MRC Clinical Sciences Centre Imperial College London Hammersmith Hospital Campus Du Cane Road London W12 0NN United Kingdom Tel: +44-20-8383 8285
_______________________________________________ galaxy-user mailing list galaxy-user@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-user

Hi Jeremy, I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads): 1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV. What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings. Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere? A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem. Best Wishes, David. __________________________________ Dr David A. Matthews Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K. Tel. +44 117 3312058 Fax. +44 117 3312091 D.A.Matthews@bristol.ac.uk
Hi all, Further to my last email, I've published a workflow (Bristol workflow ....) which does what I described below - hope this helps in understanding what I'm on about (!). Best Wishes, David. On 23 Feb 2011, at 14:41, David Matthews wrote:
Hi Jeremy,
I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads):
1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV.
What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings.
Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere?
A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Hi David, This is a really interesting workflow. My comments: (1) I encourage you to start a discussion about this idea on seqanswers.com; you'll reach more people and may have a better discussion there. Ideally, you'll get a Tophat developer to chime in on what I perceive to be the main issue, which is:
This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read.
Remember that Tophat uses Bowtie to map reads, so it would make sense to look carefully at the Bowtie documentation to see how it handles paired-end reads. I can't find anything that directly addresses your issue. The other thing to consider is how Tophat maps reads -- it breaks them up in order to find splice junctions -- and so I'm not sure that Tophat/Bowtie is really mapping paired reads; it may be doing some hybrid single/paired-end mapping. Also, at one time, you could specify Bowtie parameters when running Tophat, but I don't see that option anymore. (2) It would be interesting to know whether you get qualitatively different results via Cufflinks (or another transcriptome analysis software package) using your method vs. just using Tophat w/ and w/o ignoring non-unique reads. A skeptical view of your workflow would note that (a) multi-mapping reads may be legitimate and should not be filtered out and (b) Cufflinks/compare/diff assembly and quantitation may smooth out stray reads enough so that your method isn't necessary. Thanks for the interesting post, J. On Feb 23, 2011, at 9:41 AM, David Matthews wrote:
Hi Jeremy,
I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads):
1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV.
What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings.
Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere?
A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk

David, Just curious are you using latest version of TopHat ( 1.2.0 released on 01/18/11). I was in contact with TopHat team about similar issue of paired end reads. They suggested that this concern has been addressed in new version. I have to confirm the claim with my run. Vasu Punj --- On Wed, 2/23/11, Jeremy Goecks <jeremy.goecks@emory.edu> wrote: From: Jeremy Goecks <jeremy.goecks@emory.edu> Subject: Re: [galaxy-user] RNA seq analysis To: "David Matthews" <d.a.matthews@bristol.ac.uk> Cc: galaxy-user@bx.psu.edu Date: Wednesday, February 23, 2011, 10:05 PM Hi David, This is a really interesting workflow. My comments: (1) I encourage you to start a discussion about this idea on seqanswers.com; you'll reach more people and may have a better discussion there. Ideally, you'll get a Tophat developer to chime in on what I perceive to be the main issue, which is: This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. Remember that Tophat uses Bowtie to map reads, so it would make sense to look carefully at the Bowtie documentation to see how it handles paired-end reads. I can't find anything that directly addresses your issue. The other thing to consider is how Tophat maps reads -- it breaks them up in order to find splice junctions -- and so I'm not sure that Tophat/Bowtie is really mapping paired reads; it may be doing some hybrid single/paired-end mapping. Also, at one time, you could specify Bowtie parameters when running Tophat, but I don't see that option anymore. (2) It would be interesting to know whether you get qualitatively different results via Cufflinks (or another transcriptome analysis software package) using your method vs. just using Tophat w/ and w/o ignoring non-unique reads. A skeptical view of your workflow would note that (a) multi-mapping reads may be legitimate and should not be filtered out and (b) Cufflinks/compare/diff assembly and quantitation may smooth out stray reads enough so that your method isn't necessary. Thanks for the interesting post, J. On Feb 23, 2011, at 9:41 AM, David Matthews wrote: Hi Jeremy, I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads): 1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV. What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings. Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere? A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem. Best Wishes, David. __________________________________ Dr David A. Matthews Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K. Tel. +44 117 3312058 Fax. +44 117 3312091 D.A.Matthews@bristol.ac.uk -----Inline Attachment Follows----- _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/

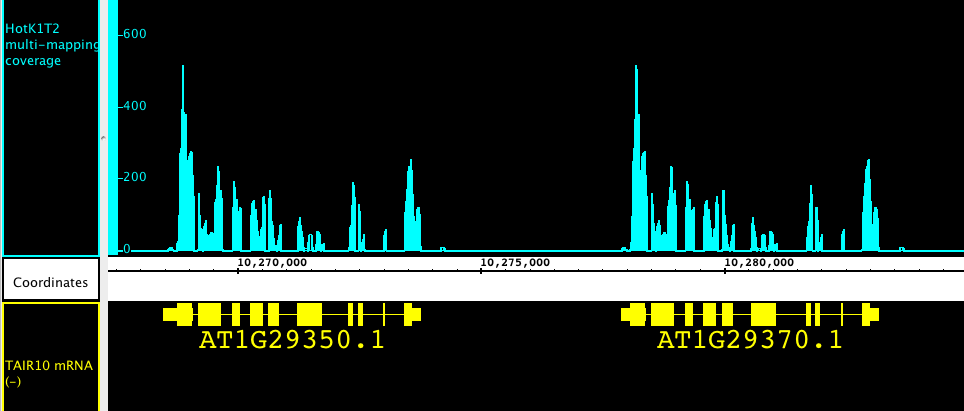
Hello, I like your approach of running the alignment tools with liberal settings and then filtering the results into different categories. This discussion reminds me of how in expression microarray analysis, we face uncertainty as to what molecules (exactly) are hybridizing to the probes on a chip. Maybe the ambiguity of mapping short sequence reads introduces similar uncertainty? I also like your idea of capturing the reads that map multiple times. It¹s interesting to visualize the alignments for reads that map onto multiple locations in a genome. An example (from data expressed in ³wiggle² format) is described here: https://wiki.transvar.org/confluence/x/w4BJAQ My apologies for posting another IGB citation, but I think it can be interesting and informative to see the data in this way, and IGB makes it easy to zoom in and out through the data and find patterns quickly. One of the first things I noticed when I started looking at coverage graphs made from multi-mapping reads is that (1) there are a lot of them and (2) they expose tandemly duplicated genes. I attach an image that shows a particularly striking example from a single-read, 75 bp RNA-Seq data set from Arabidopsis thaliana Col-0. The pattern of read alignment is nearly identical between the two genes. You can¹t see it from the image, of course, but if I right-click one of the genes, IGB links out to a Web page describing the gene at www.arabidopsis.org, the main on-line database for Arabidopsis. (Human genes link to NCBI.) -Ann On 2/23/11 11:05 PM, "Jeremy Goecks" <jeremy.goecks@emory.edu> wrote:
Hi David,
This is a really interesting workflow. My comments:
(1) I encourage you to start a discussion about this idea on seqanswers.com <http://seqanswers.com> ; you'll reach more people and may have a better discussion there. Ideally, you'll get a Tophat developer to chime in on what I perceive to be the main issue, which is:
This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read.
Remember that Tophat uses Bowtie to map reads, so it would make sense to look carefully at the Bowtie documentation to see how it handles paired-end reads. I can't find anything that directly addresses your issue. The other thing to consider is how Tophat maps reads -- it breaks them up in order to find splice junctions -- and so I'm not sure that Tophat/Bowtie is really mapping paired reads; it may be doing some hybrid single/paired-end mapping. Also, at one time, you could specify Bowtie parameters when running Tophat, but I don't see that option anymore.
(2) It would be interesting to know whether you get qualitatively different results via Cufflinks (or another transcriptome analysis software package) using your method vs. just using Tophat w/ and w/o ignoring non-unique reads. A skeptical view of your workflow would note that (a) multi-mapping reads may be legitimate and should not be filtered out and (b) Cufflinks/compare/diff assembly and quantitation may smooth out stray reads enough so that your method isn't necessary.
Thanks for the interesting post, J.
On Feb 23, 2011, at 9:41 AM, David Matthews wrote:
Hi Jeremy,
I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads):
1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV.
What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings.
Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere?
A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org
{kind=link}
Hello, I like your approach of running the alignment tools with liberal settings and then filtering the results into different categories. This discussion reminds me of how in expression microarray analysis, we face uncertainty as to what molecules (exactly) are hybridizing to the probes on a chip. Maybe the ambiguity of mapping short sequence reads introduces similar uncertainty? I also like your idea of capturing the reads that map multiple times. It¹s interesting to visualize the alignments for reads that map onto multiple locations in a genome. An example (from data expressed in ³wiggle² format) is described here: https://wiki.transvar.org/confluence/x/w4BJAQ My apologies for posting another IGB citation, but I think it can be interesting and informative to see the data in this way, and IGB makes it easy to zoom in and out through the data and find patterns quickly. One of the first things I noticed when I started looking at coverage graphs made from multi-mapping reads is that (1) there are a lot of them and (2) they expose tandemly duplicated genes. Here¹s a link to an image that showing a particularly striking example from a single-read, 75 bp RNA-Seq data set from Arabidopsis thaliana Col-0. The pattern of read alignment is nearly identical between the two genes. https://wiki.transvar.org/confluence/download/attachments/21594307/tandem-du plication.png You can¹t see it from the image, of course, but if I right-click one of the genes, IGB links out to a Web page describing the gene at www.arabidopsis.org, the main on-line database for Arabidopsis. (Human genes link to NCBI.) -Ann On 2/23/11 11:05 PM, "Jeremy Goecks" <jeremy.goecks@emory.edu> wrote:
Hi David,
This is a really interesting workflow. My comments:
(1) I encourage you to start a discussion about this idea on seqanswers.com <http://seqanswers.com> ; you'll reach more people and may have a better discussion there. Ideally, you'll get a Tophat developer to chime in on what I perceive to be the main issue, which is:
This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read.
Remember that Tophat uses Bowtie to map reads, so it would make sense to look carefully at the Bowtie documentation to see how it handles paired-end reads. I can't find anything that directly addresses your issue. The other thing to consider is how Tophat maps reads -- it breaks them up in order to find splice junctions -- and so I'm not sure that Tophat/Bowtie is really mapping paired reads; it may be doing some hybrid single/paired-end mapping. Also, at one time, you could specify Bowtie parameters when running Tophat, but I don't see that option anymore.
(2) It would be interesting to know whether you get qualitatively different results via Cufflinks (or another transcriptome analysis software package) using your method vs. just using Tophat w/ and w/o ignoring non-unique reads. A skeptical view of your workflow would note that (a) multi-mapping reads may be legitimate and should not be filtered out and (b) Cufflinks/compare/diff assembly and quantitation may smooth out stray reads enough so that your method isn't necessary.
Thanks for the interesting post, J.
On Feb 23, 2011, at 9:41 AM, David Matthews wrote:
Hi Jeremy,
I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads):
1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV.
What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings.
Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere?
A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org
Thanks Ann for your comments and for the stuff you showed at IGB - looks very interesting. I agree that multihits may the equivalent of the problem you describe from microarrays. I think, for me anyway, knowing the scale if the issue is the key thing at this stage. As you imply from your email the next -and potentially very interesting step - is to figure out how/where these multihits are and how they came to be. I guess it all comes dow to where do genes come from? Well, many of them come from other genes via duplications, transpositions etc etc! I have made a slight alteration to this "bristol" workflow which now automatically creates a sorted sam file of the multihits (forgot to put it in 1st time round!) Cheers David On 24 Feb 2011, at 12:08, Ann Loraine wrote:
Hello,
I like your approach of running the alignment tools with liberal settings and then filtering the results into different categories.
This discussion reminds me of how in expression microarray analysis, we face uncertainty as to what molecules (exactly) are hybridizing to the probes on a chip.
Maybe the ambiguity of mapping short sequence reads introduces similar uncertainty?
I also like your idea of capturing the reads that map multiple times.
It’s interesting to visualize the alignments for reads that map onto multiple locations in a genome.
An example (from data expressed in “wiggle” format) is described here:
https://wiki.transvar.org/confluence/x/w4BJAQ
My apologies for posting another IGB citation, but I think it can be interesting and informative to see the data in this way, and IGB makes it easy to zoom in and out through the data and find patterns quickly.
One of the first things I noticed when I started looking at coverage graphs made from multi-mapping reads is that (1) there are a lot of them and (2) they expose tandemly duplicated genes.
Here’s a link to an image that showing a particularly striking example from a single-read, 75 bp RNA-Seq data set from Arabidopsis thaliana Col-0. The pattern of read alignment is nearly identical between the two genes. https://wiki.transvar.org/confluence/download/attachments/21594307/tandem-du...
You can’t see it from the image, of course, but if I right-click one of the genes, IGB links out to a Web page describing the gene at www.arabidopsis.org, the main on-line database for Arabidopsis. (Human genes link to NCBI.)
-Ann
On 2/23/11 11:05 PM, "Jeremy Goecks" <jeremy.goecks@emory.edu> wrote:
Hi David,
This is a really interesting workflow. My comments:
(1) I encourage you to start a discussion about this idea on seqanswers.com <http://seqanswers.com> ; you'll reach more people and may have a better discussion there. Ideally, you'll get a Tophat developer to chime in on what I perceive to be the main issue, which is:
This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read.
Remember that Tophat uses Bowtie to map reads, so it would make sense to look carefully at the Bowtie documentation to see how it handles paired-end reads. I can't find anything that directly addresses your issue. The other thing to consider is how Tophat maps reads -- it breaks them up in order to find splice junctions -- and so I'm not sure that Tophat/Bowtie is really mapping paired reads; it may be doing some hybrid single/paired-end mapping. Also, at one time, you could specify Bowtie parameters when running Tophat, but I don't see that option anymore.
(2) It would be interesting to know whether you get qualitatively different results via Cufflinks (or another transcriptome analysis software package) using your method vs. just using Tophat w/ and w/o ignoring non-unique reads. A skeptical view of your workflow would note that (a) multi-mapping reads may be legitimate and should not be filtered out and (b) Cufflinks/compare/diff assembly and quantitation may smooth out stray reads enough so that your method isn't necessary.
Thanks for the interesting post, J.
On Feb 23, 2011, at 9:41 AM, David Matthews wrote:
Hi Jeremy,
I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads):
1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV.
What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings.
Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere?
A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
HI Jeremy, Thanks for the feedback. I know what you mean about tophat not having the same functionality of bowtie. However, I think whatever tophat does do (now or in the future) I think it is useful to collect the multihits separately since either you leave them in and over estimate gene expression or remove them and underestimate gene expression. As you suggested I put this up on Seqanswers to see if anyone else likes/doesn't like it we'll see how it goes. I certainly find it handy - not least to reassure myself that when I get the gene expresison data I can tell if there are any "funny" reads making up the numbers! Cheers David P.S. I modified the workflow to include collecting the multihits in a separate sorted sam file. On 24 Feb 2011, at 04:05, Jeremy Goecks wrote:
Hi David,
This is a really interesting workflow. My comments:
(1) I encourage you to start a discussion about this idea on seqanswers.com; you'll reach more people and may have a better discussion there. Ideally, you'll get a Tophat developer to chime in on what I perceive to be the main issue, which is:
This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read.
Remember that Tophat uses Bowtie to map reads, so it would make sense to look carefully at the Bowtie documentation to see how it handles paired-end reads. I can't find anything that directly addresses your issue. The other thing to consider is how Tophat maps reads -- it breaks them up in order to find splice junctions -- and so I'm not sure that Tophat/Bowtie is really mapping paired reads; it may be doing some hybrid single/paired-end mapping. Also, at one time, you could specify Bowtie parameters when running Tophat, but I don't see that option anymore.
(2) It would be interesting to know whether you get qualitatively different results via Cufflinks (or another transcriptome analysis software package) using your method vs. just using Tophat w/ and w/o ignoring non-unique reads. A skeptical view of your workflow would note that (a) multi-mapping reads may be legitimate and should not be filtered out and (b) Cufflinks/compare/diff assembly and quantitation may smooth out stray reads enough so that your method isn't necessary.
Thanks for the interesting post, J.
On Feb 23, 2011, at 9:41 AM, David Matthews wrote:
Hi Jeremy,
I thought I'd write to get a discussion of a workflow for people doing RNA seq that I have found very useful and addresses some issues in mapping mRNA derived RNA-seq paired end data to the genome using tophat. Here is the approach I use (I have a human mRNA sample deep sequenced with a 56bp paired end read on an illumina generating 29 million reads):
1. Align to hg19 (in my case) using tophat and allowing up to 40 hits for each sequence read 2. In samtools filter for "read is unmapped", "mate is mapped" and "mate is mapped in a proper pair" 3. Use "group" to group the filtered sam file on c1 (which is the "bio-sequencer" read number) and set an operation to count on c1 as well. This provides a list of the reads and how many times they map to the human genome, because you have filtered the set for reads that have a mate pair there will be an even number for each read. For most of the reads the number will be 2 (indicating the forward read maps once and the reverse read maps once and in a proper pair) but for reads that map ambiguously the number will be multiples of 2. If you count these up I find that 18 million reads map once, 1.3 million map twice, 400,000 reads map 3 times and so on until you get down to 1 read mapping 30 times, 1 read mapping 31 times and so on... 4. Filter the reads to remove any reads that map more than 2 times. 5. Use "compare two datasets" to compare your new list of reads that map only twice to pull out all the reads in your sam file that only map twice (i.e. the mate pairs). 6. You'll need to sort the sam file before you can use it with other applications like IGV.
What you end up with is a sam file where all the reads map to one site only and all the reads map as a proper pair. This may seem similar to setting tophat to ignore non-unique reads. However, it is not. This approach gives you 10-15% more reads. I think it is because if tophat finds (for example) that the forward read maps to one site but the reverse read maps to two sites it throws away the whole read. By filtering the sam file to restrict it to only those mappings that make sense you increase the number of unique reads by getting rid of irrational mappings.
Has anyone else found this? Does this make sense to anyone else? Am I making a huge mistake somewhere?
A nice aspect of this (or at least I think so!) is that by filtering in this manner you can also create a sam file of non-unique mappings which you can monitor. This can be useful if one or more genes has a problem of generating a lot of non-unique maps which may give problems accurately estimating its expression. Also, you also get a list of how many multi hits you have in your data so you know the scale of the problem.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
participants (6)
-
 Ann Loraine
Ann Loraine -
 David Matthews
David Matthews -
David Matthews
-
 Jeremy Goecks
Jeremy Goecks -
 Motallebipour, Mehdi
Motallebipour, Mehdi -
 vasu punj
vasu punj