unable to import run or save-to-file published workflow after galaxy upgrade

dear all, we've just upgraded our Galaxy server (Galaxy revision 7148:17d57db9a7c0, upgraded to revision 10422:a886bc3ae924 ), and have found that an NGS training workflow that one of us set up and published is no longer accessible - we now get a stack trace when we try to run it (screenshot attached) ; if we try to save to file, we "get page not found" from the browser, but no errors reported in web logs, or Galaxy log as far as we can see Is there any way we can recover the workflow ? How to avoid this happening in future upgrades ? This time it is not a major problem, however we would not want this to happen to production workflows. Should we save-to-file on all workflows as a precaution before an upgrade ? (Would that help ? ) The workflow included steps as below. Grateful for any suggestions. Cheers Alan McC First Part : checking GC content *Upload the sampling.fasta file or get it from the shared data in Galaxy. *Use geecee from EMBOSS *Remove beginning (Text manipulation), first line *Convert as tabular (Text manipulation) or cut (Text manipulation) *Histogram (Graph/Display Data)on col2 *Compute data (Text manipulation) (data4 convert; col2>0.2) *Count data (statistics on col3) If it is worth it we can remove the sequences which have a too low GC content 3 Second part : sequence length *Compute Sequence length (FASTA manipulation) *Summary Statistics (Statistics)on col2 *Filter by length(FASTA manipulation) ; 800 *Line/world/character count(Text manipulation) *Blastn against fungi db : yeast, only one hit to show (advanced options ) *Count the lines to know how many sequences have a hit . (Text manipulation) 4 Bonus track : removing sequences with low GC content *Select (filter and sort) on data 6 where matching = True *FASTA-to-Tabular (FASTA manipulation) the input file sampling.fasta (2 col for the title) *Join two datasets(Join, Subtract and Group) on the columns c1 *Tabular-to-FASTA (fasta manipulation) of the previous results. *You can see how many sequences you took away (click on the dataset name in History) and it should correspond to the number of True in Count . 5
{kind=link}

Moved to galaxy-dev@bx.psu.edu On 10/8/13 4:44 PM, McCulloch, Alan wrote:
dear all,
we've just upgraded our Galaxy server (Galaxy revision 7148:17d57db9a7c0, upgraded to revision 10422:a886bc3ae924 ), and
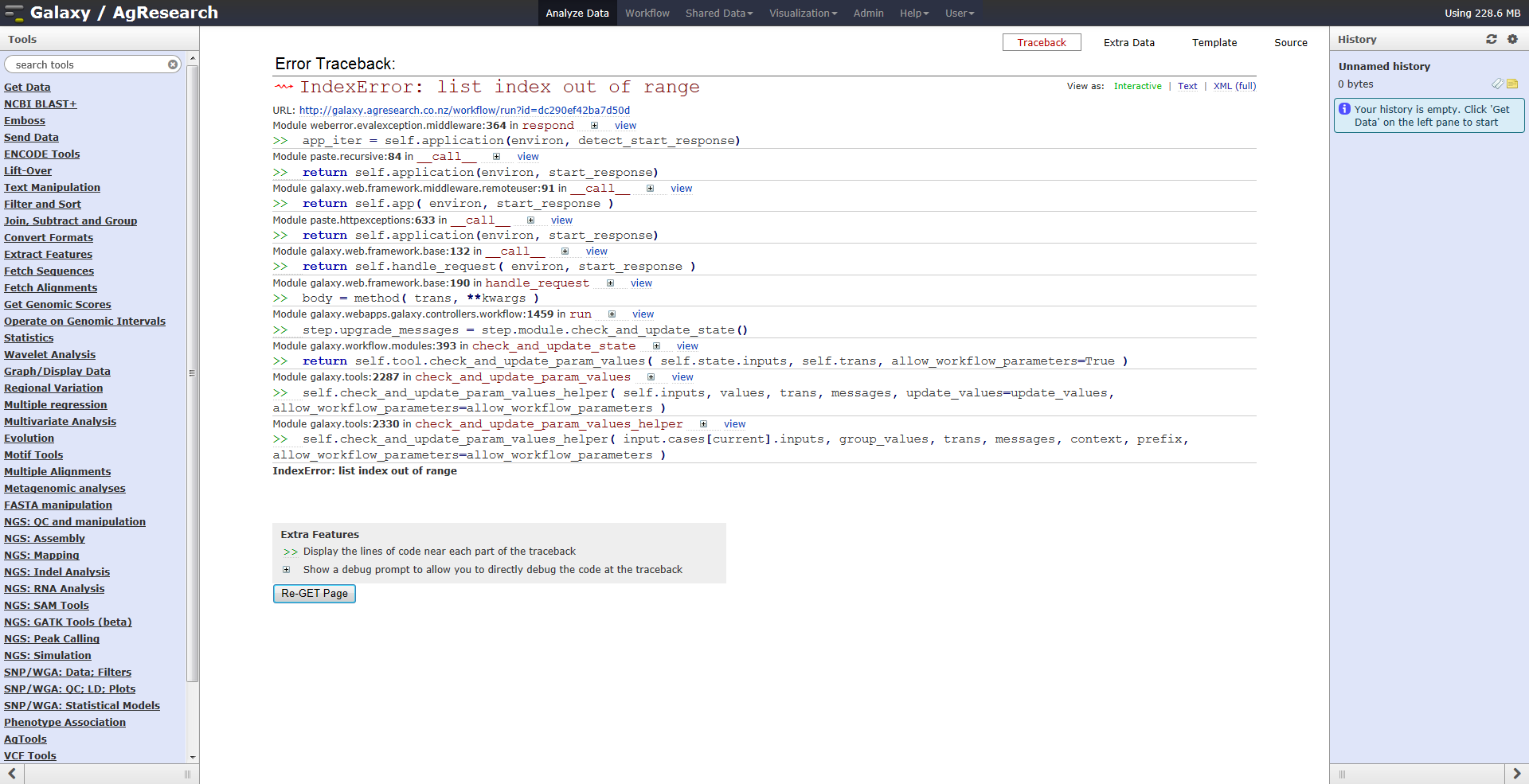
have found that an NGS training workflow that one of us set up and published is no longer accessible - we now get a stack trace
when we try to run it (screenshot attached) ; if we try to save to file, we "get page not found" from the browser, but no errors
reported in web logs, or Galaxy log as far as we can see
Is there any way we can recover the workflow ?
How to avoid this happening in future upgrades ? This time it is not a major problem, however we would not want this to happen to
production workflows. Should we save-to-file on all workflows as a precaution before an upgrade ? (Would that help ? )
The workflow included steps as below.
Grateful for any suggestions.
Cheers
Alan McC
First Part : checking GC content
.Upload the sampling.fasta file or get it from the shared data in Galaxy.
.Use geecee from EMBOSS
.Remove beginning (Text manipulation), first line
.Convert as tabular (Text manipulation) or cut (Text manipulation)
.Histogram (Graph/Display Data)on col2
.Compute data (Text manipulation) (data4 convert; col2>0.2)
.Count data (statistics on col3) If it is worth it we can remove the sequences which have a too low GC content
3
Second part : sequence length
.Compute Sequence length (FASTA manipulation)
.Summary Statistics (Statistics)on col2
.Filter by length(FASTA manipulation) ; 800
.Line/world/character count(Text manipulation)
.Blastn against fungi db : yeast, only one hit to show (advanced options )
.Count the lines to know how many sequences have a hit . (Text manipulation)
4
Bonus track : removing sequences with low GC content
.Select (filter and sort) on data 6 where matching = True
.FASTA-to-Tabular (FASTA manipulation) the input file sampling.fasta (2 col for the title)
.Join two datasets(Join, Subtract and Group) on the columns c1
.Tabular-to-FASTA (fasta manipulation) of the previous results.
.You can see how many sequences you took away (click on the dataset name in History) and it should correspond to the number of True in Count .
5
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
To search Galaxy mailing lists use the unified search at:
-- Jennifer Hillman-Jackson http://galaxyproject.org

Is there any way we can recover the workflow ?
Can the workflow's owner view and edit the workflow? My best guess is that you'll need to reinstall some tools that were migrated from the Galaxy framework to the toolshed. See here for details: http://wiki.galaxyproject.org/MigratingToolsFromGalaxyDistribution
J.
participants (3)
-
 Jennifer Jackson
Jennifer Jackson -
 Jeremy Goecks
Jeremy Goecks -
 McCulloch, Alan
McCulloch, Alan