Re: [galaxy-user] FASTQ type change

Steve, I'm sending this question along to the galaxy-user list because it's likely of interest to others as well as yourself. Dannon, our resident workflow expert, should be able to answer your question. Thanks, J. On Feb 9, 2011, at 10:28 AM, Stephen Taylor wrote:
Hi,
I seem to remember sending a question about how to change the type of fastq in a workflow to the galaxy-dev list so it can run vs bowtie without going through fastqgroomer. I think you replied to me about a tool that would do this but I can't seem to find the answer on the list or in my inbox? Is there a way to to do this or did I dream the question/answer?!:-)
Kind regards and thanks,
Steve

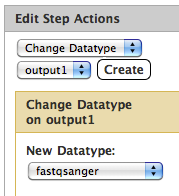
If you're confident about the format of the dataset in question, add a Change Datatype action to your workflow (on the step before bowtie) that sets the datatype to fastqsanger. See the right panel of the workflow editor under Step Actions; you want something like this: -Dannon On Feb 9, 2011, at 10:58 AM, Jeremy Goecks wrote:
Steve,
I'm sending this question along to the galaxy-user list because it's likely of interest to others as well as yourself. Dannon, our resident workflow expert, should be able to answer your question.
Thanks, J.
On Feb 9, 2011, at 10:28 AM, Stephen Taylor wrote:
Hi,
I seem to remember sending a question about how to change the type of fastq in a workflow to the galaxy-dev list so it can run vs bowtie without going through fastqgroomer. I think you replied to me about a tool that would do this but I can't seem to find the answer on the list or in my inbox? Is there a way to to do this or did I dream the question/answer?!:-)
Kind regards and thanks,
Steve
_______________________________________________ galaxy-user mailing list galaxy-user@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-user
{kind=link}

On 09/02/2011 16:18, Dannon Baker wrote:
If you're confident about the format of the dataset in question, add a Change Datatype action to your workflow (on the step before bowtie) that sets the datatype to fastqsanger.
See the right panel of the workflow editor under Step Actions; you want something like this:
Thanks. What if (after upload) this is your first step? With 'Upload' you don't get the option to do 'Change Datatype'. Steve
On Feb 9, 2011, at 12:47 PM, Stephen Taylor wrote:
Thanks.
What if (after upload) this is your first step? With 'Upload' you don't get the option to do 'Change Datatype'.
I'm not exactly sure I follow. If the first step in your workflow after the Input Dataset step is Bowtie, then you won't want to do this in the workflow, it should happen prior to that. The workflow expects that you're supplying the correct format for that first step. When you are first uploading the file you can specify at that time that it should be a fastqsanger file. For datasets that are already in a history, you can click the pencil icon on the history item and change the datatype there without running the fastq groomer. Let me know if I've misunderstood and this doesn't cover what you're looking to do. Thanks! -Dannon
On 09/02/2011 19:09, Dannon Baker wrote:
On Feb 9, 2011, at 12:47 PM, Stephen Taylor wrote:
Thanks.
What if (after upload) this is your first step? With 'Upload' you don't get the option to do 'Change Datatype'.
I'm not exactly sure I follow. If the first step in your workflow after the Input Dataset step is Bowtie, then you won't want to do this in the workflow, it should happen prior to that. The workflow expects that you're supplying the correct format for that first step.
When you are first uploading the file you can specify at that time that it should be a fastqsanger file. For datasets that are already in a history, you can click the pencil icon on the history item and change the datatype there without running the fastq groomer.
Let me know if I've misunderstood and this doesn't cover what you're looking to do.
I think you have but it doesn't help. :-) The issue is we get a lot of Illumina 1.3+ format FASTQ files and bowtie in Galaxy by default doesn't accept them although there is an option on the command line bowtie to read these. So I think the solution seems to be either hardwire the code in our local Galaxy instance to use the --solexa1.3-quals option or (probably more useful) put a drop down list in the web UI to allow the user to set the format of the fastq sequences on the bowtie tool. Thanks for you help, Steve

On Thu, Feb 10, 2011 at 12:49 PM, Stephen Taylor <stephen.taylor@imm.ox.ac.uk> wrote:
I think you have but it doesn't help. :-)
The issue is we get a lot of Illumina 1.3+ format FASTQ files and bowtie in Galaxy by default doesn't accept them although there is an option on the command line bowtie to read these. So I think the solution seems to be either hardwire the code in our local Galaxy instance to use the --solexa1.3-quals option or (probably more useful) put a drop down list in the web UI to allow the user to set the format of the fastq sequences on the bowtie tool.
Not the best approach. I think you should update the XML to include the --solexa1.3-quals option if the Galaxy file format is fastqillumina, see for example (in the reverse situation) the -Q 33 option is only used on fastqsanger when calling the FASTX tools, e.g. https://bitbucket.org/galaxy/galaxy-central/src/default/tools/fastx_toolkit/... That way the user must mark their FASTQ file type as usual (at upload time or via the "pencil icon" to edit the attributes), and then bowtie will be called appropriately. Peter
On 10/02/2011 13:05, Peter Cock wrote:
On Thu, Feb 10, 2011 at 12:49 PM, Stephen Taylor <stephen.taylor@imm.ox.ac.uk> wrote:
I think you have but it doesn't help. :-)
The issue is we get a lot of Illumina 1.3+ format FASTQ files and bowtie in Galaxy by default doesn't accept them although there is an option on the command line bowtie to read these. So I think the solution seems to be either hardwire the code in our local Galaxy instance to use the --solexa1.3-quals option or (probably more useful) put a drop down list in the web UI to allow the user to set the format of the fastq sequences on the bowtie tool.
Not the best approach.
I think you should update the XML to include the --solexa1.3-quals option if the Galaxy file format is fastqillumina, see for example (in the reverse situation) the -Q 33 option is only used on fastqsanger when calling the FASTX tools, e.g.
https://bitbucket.org/galaxy/galaxy-central/src/default/tools/fastx_toolkit/...
That way the user must mark their FASTQ file type as usual (at upload time or via the "pencil icon" to edit the attributes), and then bowtie will be called appropriately.
Ok. Cool. I didn't realise you could do that! Sounds like this should be added into the main release. It would save a lot of time/disk space instead of using Groomer. Thanks, Steve
On Thu, Feb 10, 2011 at 6:34 PM, Stephen Taylor <stephen.taylor@imm.ox.ac.uk> wrote:
On 10/02/2011 13:05, Peter Cock wrote:
On Thu, Feb 10, 2011 at 12:49 PM, Stephen Taylor <stephen.taylor@imm.ox.ac.uk> wrote:
I think you have but it doesn't help. :-)
The issue is we get a lot of Illumina 1.3+ format FASTQ files and bowtie in Galaxy by default doesn't accept them although there is an option on the command line bowtie to read these. So I think the solution seems to be either hardwire the code in our local Galaxy instance to use the --solexa1.3-quals option or (probably more useful) put a drop down list in the web UI to allow the user to set the format of the fastq sequences on the bowtie tool.
Not the best approach.
I think you should update the XML to include the --solexa1.3-quals option if the Galaxy file format is fastqillumina, see for example (in the reverse situation) the -Q 33 option is only used on fastqsanger when calling the FASTX tools, e.g.
https://bitbucket.org/galaxy/galaxy-central/src/default/tools/fastx_toolkit/...
That way the user must mark their FASTQ file type as usual (at upload time or via the "pencil icon" to edit the attributes), and then bowtie will be called appropriately.
Ok. Cool. I didn't realise you could do that!
Sounds like this should be added into the main release. It would save a lot of time/disk space instead of using Groomer.
I agree. Maybe Dan can take care of it - I don't have bowtie setup on our local Galaxy (yet) so I wouldn't be able to test the proposed fix. In the long term however the Solexa/Illumina FASTQ formats are on their way out since CASAVA 1.8 will switch to the Sanger FASTQ encoding: http://seqanswers.com/forums/showthread.php?t=8895 Peter
On Thu, Feb 10, 2011 at 6:42 PM, Peter wrote:
On Thu, Feb 10, 2011 at 6:34 PM, Stephen Taylor wrote:
Sounds like this should be added into the main release. It would save a lot of time/disk space instead of using Groomer.
I agree. Maybe Dan can take care of it - I don't have bowtie setup on our local Galaxy (yet) so I wouldn't be able to test the proposed fix.
In the long term however the Solexa/Illumina FASTQ formats are on their way out since CASAVA 1.8 will switch to the Sanger FASTQ encoding: http://seqanswers.com/forums/showthread.php?t=8895
Hi Stephen, Maybe you should file an issue on bitbucket about extending the Bowtie wrapper to support fastqillumina as well as fastqsanger as input? https://bitbucket.org/galaxy/galaxy-central/issues?status=new&status=open The current version of the wrapper is here if you want to look at it: https://bitbucket.org/galaxy/galaxy-central/src/default/tools/sr_mapping/bow... Peter

Hi Stephen and Peter, This change is definitely possible, and because the SAM format specifies that its quality scores are phred scaled and ASCII offset of 33 (regardless of provided input) it shouldn't cause complications downstream. We'll add this to our todo list. I created a ticket: https://bitbucket.org/galaxy/galaxy-central/issue/471/allow-bowtie-mapper-to... that you can follow if you like. Thanks, Dan On Feb 10, 2011, at 1:42 PM, Peter Cock wrote:
On Thu, Feb 10, 2011 at 6:34 PM, Stephen Taylor <stephen.taylor@imm.ox.ac.uk> wrote:
On 10/02/2011 13:05, Peter Cock wrote:
On Thu, Feb 10, 2011 at 12:49 PM, Stephen Taylor <stephen.taylor@imm.ox.ac.uk> wrote:
I think you have but it doesn't help. :-)
The issue is we get a lot of Illumina 1.3+ format FASTQ files and bowtie in Galaxy by default doesn't accept them although there is an option on the command line bowtie to read these. So I think the solution seems to be either hardwire the code in our local Galaxy instance to use the --solexa1.3-quals option or (probably more useful) put a drop down list in the web UI to allow the user to set the format of the fastq sequences on the bowtie tool.
Not the best approach.
I think you should update the XML to include the --solexa1.3-quals option if the Galaxy file format is fastqillumina, see for example (in the reverse situation) the -Q 33 option is only used on fastqsanger when calling the FASTX tools, e.g.
https://bitbucket.org/galaxy/galaxy-central/src/default/tools/fastx_toolkit/...
That way the user must mark their FASTQ file type as usual (at upload time or via the "pencil icon" to edit the attributes), and then bowtie will be called appropriately.
Ok. Cool. I didn't realise you could do that!
Sounds like this should be added into the main release. It would save a lot of time/disk space instead of using Groomer.
I agree. Maybe Dan can take care of it - I don't have bowtie setup on our local Galaxy (yet) so I wouldn't be able to test the proposed fix.
In the long term however the Solexa/Illumina FASTQ formats are on their way out since CASAVA 1.8 will switch to the Sanger FASTQ encoding: http://seqanswers.com/forums/showthread.php?t=8895
Peter
_______________________________________________ galaxy-user mailing list galaxy-user@lists.bx.psu.edu http://lists.bx.psu.edu/listinfo/galaxy-user
participants (5)
-
 Daniel Blankenberg
Daniel Blankenberg -
 Dannon Baker
Dannon Baker -
 Jeremy Goecks
Jeremy Goecks -
 Peter Cock
Peter Cock -
 Stephen Taylor
Stephen Taylor