GFF not recognized in CUFFLINK

Dear Team, I've been having a problem with cufflink regarding GFF files. I tried searching the mailing list first and failed to find an answer. Could you help me look at this? I downloaded my genome annotation GFF file from NCBI (soon I realized NCBI format may be a problem) for my bacterial RNA-seq data analysis. My GFF file looks like the following: '##gff-version 3#!gff-spec-version 1.20#!processor NCBI annotwriter##sequence-region NC_011420.2 1 4355543##species http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684NC_011420.2 RefSeqregion14355543.+.ID=id0;Dbxref=taxon:414684;Is_circular=true;culture-collection=ATCC:51521;gb-synonym=Rhodocista centenaria SW;gbkey=Src;genome=chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521NC_011420.2RefSeqgene113343.+. ID=gene0;Name=RC1_0011;Dbxref=GeneID:7008893;gbkey=Gene;locus_tag=RC1_0011 NC_011420.2RefSeqCDS113343.+0ID=cds0;Name=YP_002296275.1;Parent=gene0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP_002296275.1,GeneID:7008893;gbkey=CDS;product=hypothetical protein;protein_id=YP_002296275.1;transl_table=11 I used this file for cufflink but all the FPKM values are 0. I checked out this link: http://cufflinks.cbcb.umd.edu/gff.html and thought that maybe the problem is because I don't have any mRNA feature in my gff file. Since I am dealing with a bacterial genome, there is no exon/intron or UTR info needed. Therefore I modified my GFF file into the following: ##gff-version 3#!gff-spec-version 1.20#!processor NCBI annotwriter##sequence-region NC_011420.2 1 4355543##species http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684NC_011420.2 RefSeqregion14355543.+.ID=id0;Dbxref=taxon:414684;Is_circular=true;culture-collection=ATCC:51521;gb-synonym=Rhodocista centenaria SW;gbkey=Src;genome=chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521NC_011420.2RefSeqmRNA113343.+. ID=mRNA0;Name=RC1_0011;Dbxref=GeneID:7008893;gbkey=Gene;locus_tag=RC1_0011 NC_011420.2RefSeqCDS113343.+0ID=cds0;Name=YP_002296275.1;Parent=mRNA0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP_002296275.1,GeneID:7008893;gbkey=CDS;product=hypothetical protein;protein_id=YP_002296275.1;transl_table=11 I re-ran cufflink however this time there is error reported. I can only tell from the report that there is a segmentation fault but not further details. The report is as follows: Error running cufflinks. return code = 139 Command line: cufflinks -q --no-update-check -I 100 -F 0.100000 -j 0.150000 -p 4 -G /galaxy/test_pool/pool5/files/000/327/dataset_327777.dat /galaxy/test_database/files/000/325/dataset_325086.dat [19:41:41] Loading reference annotation. Segmentation fault cp: cannot stat `/galaxy/test_pool/pool3/tmp/job_working_directory/000/170/170197/global_model.txt': No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/job_working_directory/000/170/170197/isoforms.fpkm_tracking': No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/job_working_directory/000/170/170197/genes.fpkm_tracking': No such file or directory My questions will be: 1. Is there any way to modify a NCBI bacterial genome annotation GFF file to make it usable for cufflink? Our genome annotation is only available in NCBI, not ensemble or USDC so this is pretty much my only choice.. 2. Should I proceed with modifying the GFF file or should I convert it into GTF and use the GTF instead in cufflink? I am a biochemist and really new to the computer world so any advice will help! Thanks a lot, Qian -- Qian Dong Bauer Lab, MCBD Simon Hall: 313-317 212 S. Hawthorne Dr. Bloomington, IN 47405 Email:dong3@indiana.edu Lab Phone:812-855-8443

Hello, The first thing to double check is that the chromosome identifier is an exact match between the reference genome and the reference annotation. The GFF3 file is naming the chromosome "NC_011420.2". The reference annotation chromosome should be named exactly the same way. Check this in the input BAM/SAM datasets or the original .fasta reference genome. Hopefully this finds the problem. Correcting mismatched names (due to various reasons) is the most common solution to this sort of issue: 'Tools on the Main server: Example', bullet item #2: http://wiki.g2.bx.psu.edu/Support#Interpreting_scientific_results Best, Jen Galaxy team On 9/26/12 8:46 AM, Qian Dong wrote:
Dear Team,
I've been having a problem with cufflink regarding GFF files. I tried searching the mailing list first and failed to find an answer. Could you help me look at this?
I downloaded my genome annotation GFF file from NCBI (soon I realized NCBI format may be a problem) for my bacterial RNA-seq data analysis. My GFF file looks like the following:
' ##gff-version 3 #!gff-spec-version 1.20 #!processor NCBI annotwriter ##sequence-region NC_011420.2 1 4355543 ##species http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684 NC_011420.2 RefSeq region 1 4355543 . + . ID=id0;Dbxref=taxon:414684;Is_circular=true;culture-collection=ATCC:51521;gb-synonym=Rhodocista centenaria SW;gbkey=Src;genome=chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521 NC_011420.2 RefSeq gene 11 3343 . + . ID=gene0;Name=RC1_0011;Dbxref=GeneID:7008893;gbkey=Gene;locus_tag=RC1_0011 NC_011420.2 RefSeq CDS 11 3343 . + 0 ID=cds0;Name=YP_002296275.1;Parent=gene0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP_002296275.1,GeneID:7008893;gbkey=CDS;product=hypothetical protein;protein_id=YP_002296275.1;transl_table=11
I used this file for cufflink but all the FPKM values are 0. I checked out this link: http://cufflinks.cbcb.umd.edu/gff.html and thought that maybe the problem is because I don't have any mRNA feature in my gff file. Since I am dealing with a bacterial genome, there is no exon/intron or UTR info needed. Therefore I modified my GFF file into the following:
##gff-version 3 #!gff-spec-version 1.20 #!processor NCBI annotwriter ##sequence-region NC_011420.2 1 4355543 ##species http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684 NC_011420.2 RefSeq region 1 4355543 . + . ID=id0;Dbxref=taxon:414684;Is_circular=true;culture-collection=ATCC:51521;gb-synonym=Rhodocista centenaria SW;gbkey=Src;genome=chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521 NC_011420.2 RefSeq mRNA 11 3343 . + . ID=mRNA0;Name=RC1_0011;Dbxref=GeneID:7008893;gbkey=Gene;locus_tag=RC1_0011 NC_011420.2 RefSeq CDS 11 3343 . + 0 ID=cds0;Name=YP_002296275.1;Parent=mRNA0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP_002296275.1,GeneID:7008893;gbkey=CDS;product=hypothetical protein;protein_id=YP_002296275.1;transl_table=11
I re-ran cufflink however this time there is error reported. I can only tell from the report that there is a segmentation fault but not further details. The report is as follows:
Error running cufflinks. return code = 139 Command line: cufflinks -q --no-update-check -I 100 -F 0.100000 -j 0.150000 -p 4 -G /galaxy/test_pool/pool5/files/000/327/dataset_327777.dat /galaxy/test_database/files/000/325/dataset_325086.dat [19:41:41] Loading reference annotation. Segmentation fault
cp: cannot stat `/galaxy/test_pool/pool3/tmp/job_working_directory/000/170/170197/global_model.txt': No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/job_working_directory/000/170/170197/isoforms.fpkm_tracking': No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/job_working_directory/000/170/170197/genes.fpkm_tracking': No such file or directory
My questions will be:
1. Is there any way to modify a NCBI bacterial genome annotation GFF file to make it usable for cufflink? Our genome annotation is only available in NCBI, not ensemble or USDC so this is pretty much my only choice..
2. Should I proceed with modifying the GFF file or should I convert it into GTF and use the GTF instead in cufflink?
I am a biochemist and really new to the computer world so any advice will help!
Thanks a lot,
Qian -- Qian Dong Bauer Lab, MCBD Simon Hall: 313-317 212 S. Hawthorne Dr. Bloomington, IN 47405 Email:dong3@indiana.edu <mailto:Email%3Adong3@indiana.edu> Lab Phone:812-855-8443
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://galaxyproject.org

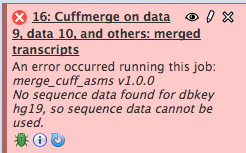
Hi, I'm getting this cuffmerge error on our local Galaxy instance. I assume it is because it doesn't have the hg19 reference genome. Can you please tell me how I would include into this Galaxy installation? Thank you.
{kind=link}
Hello, Instructions for including a new genome indexed for tools, including how to download the same indexes used on the public Main Galaxy instance hosted at http://usegalaxy.org, are in these wikis: http://wiki.g2.bx.psu.edu/Admin/Data%20Integration http://wiki.g2.bx.psu.edu/Admin/NGS%20Local%20Setup The "dbkey" that you will want to use is simply "hg19" for this genome, should you use the rsync service. Going forward, the galaxy-dev@bx.psu.edu mailing list would be a good choice for local install questions. The development community can help solve more complex problems as they come up and/or you might be interested in following discussions: http://wiki.g2.bx.psu.edu/Support#Mailing_Lists http://wiki.g2.bx.psu.edu/Mailing%20Lists Hopefully this helps! Jen Galaxy team On 9/26/12 12:47 PM, Kenneth Auerbach wrote:
Hi,
I'm getting this cuffmerge error on our local Galaxy instance. I assume it is because it doesn't have the hg19 reference genome. Can you please tell me how I would include into this Galaxy installation?
Thank you.
-- Jennifer Jackson http://galaxyproject.org
{kind=link}
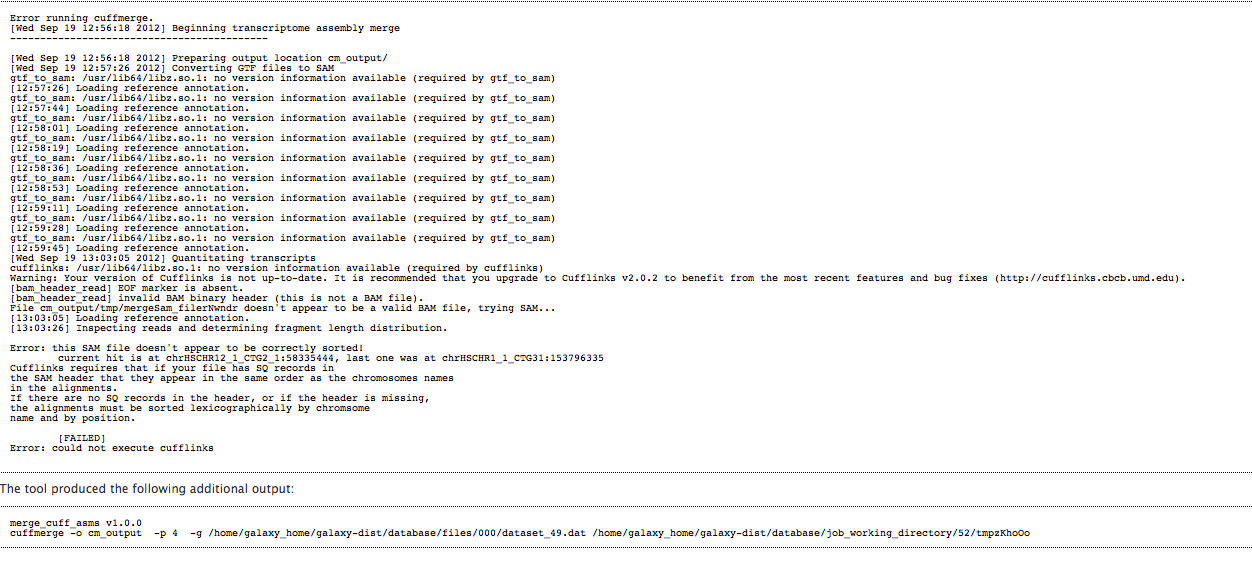
Hello, The problem is most likely with the SAM files and sorting. Please see: "Why won't my SAM dataset work with Cufflinks?" http://main.g2.bx.psu.edu/u/jeremy/p/transcriptome-analysis-faq#faq2 Best, Jen Galaxy team On 9/26/12 12:54 PM, Kenneth Auerbach wrote:
Hi,
I'm also getting these error when I run cuffmerge. It was run without a reference so it shouldn't have anything to do with the reference. Can you please tell me how to fix this?
Thank you.
-- Jennifer Jackson http://galaxyproject.org
Dear Jennifer, My .fasta reference genome is like this: '>gi|289546492|ref|NC_011420.2| Rhodospirillum centenum SW chromosome, complete genome' and in the SAM file generated by BOWTIE it says: @SQ SN:gi|289546492|ref|NC_011420.2| LN:4355543 So I think they are all the same as "NC_011420.2". Is there anything else I can try? Thank you, Qian On Wed, Sep 26, 2012 at 3:35 PM, Jennifer Jackson <jen@bx.psu.edu> wrote:
Hello,
The first thing to double check is that the chromosome identifier is an exact match between the reference genome and the reference annotation.
The GFF3 file is naming the chromosome "NC_011420.2".
The reference annotation chromosome should be named exactly the same way. Check this in the input BAM/SAM datasets or the original .fasta reference genome.
Hopefully this finds the problem. Correcting mismatched names (due to various reasons) is the most common solution to this sort of issue: 'Tools on the Main server: Example', bullet item #2: http://wiki.g2.bx.psu.edu/**Support#Interpreting_**scientific_results<http://wiki.g2.bx.psu.edu/Support#Interpreting_scientific_results>
Best,
Jen Galaxy team
On 9/26/12 8:46 AM, Qian Dong wrote:
Dear Team,
I've been having a problem with cufflink regarding GFF files. I tried searching the mailing list first and failed to find an answer. Could you help me look at this?
I downloaded my genome annotation GFF file from NCBI (soon I realized NCBI format may be a problem) for my bacterial RNA-seq data analysis. My GFF file looks like the following:
' ##gff-version 3 #!gff-spec-version 1.20 #!processor NCBI annotwriter ##sequence-region NC_011420.2 1 4355543 ##species http://www.ncbi.nlm.nih.gov/**Taxonomy/Browser/wwwtax.cgi?** id=414684<http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684> NC_011420.2 RefSeq region 1 4355543 . + . ID=id0;Dbxref=taxon:414684;Is_**circular=true;culture-** collection=ATCC:51521;gb-**synonym=Rhodocista centenaria SW;gbkey=Src;genome=**chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521 NC_011420.2 RefSeq gene 11 3343 . + . ID=gene0;Name=RC1_0011;Dbxref=**GeneID:7008893;gbkey=Gene;** locus_tag=RC1_0011 NC_011420.2 RefSeq CDS 11 3343 . + 0 ID=cds0;Name=YP_002296275.1;**Parent=gene0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP_**002296275.1,GeneID:7008893;** gbkey=CDS;product=hypothetical protein;protein_id=YP_**002296275.1;transl_table=11
I used this file for cufflink but all the FPKM values are 0. I checked out this link: http://cufflinks.cbcb.umd.edu/**gff.html<http://cufflinks.cbcb.umd.edu/gff.html>and thought that maybe the problem is because I don't have any mRNA feature in my gff file. Since I am dealing with a bacterial genome, there is no exon/intron or UTR info needed. Therefore I modified my GFF file into the following:
##gff-version 3 #!gff-spec-version 1.20 #!processor NCBI annotwriter ##sequence-region NC_011420.2 1 4355543 ##species http://www.ncbi.nlm.nih.gov/**Taxonomy/Browser/wwwtax.cgi?** id=414684<http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684> NC_011420.2 RefSeq region 1 4355543 . + . ID=id0;Dbxref=taxon:414684;Is_**circular=true;culture-** collection=ATCC:51521;gb-**synonym=Rhodocista centenaria SW;gbkey=Src;genome=**chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521 NC_011420.2 RefSeq mRNA 11 3343 . + . ID=mRNA0;Name=RC1_0011;Dbxref=**GeneID:7008893;gbkey=Gene;** locus_tag=RC1_0011 NC_011420.2 RefSeq CDS 11 3343 . + 0 ID=cds0;Name=YP_002296275.1;**Parent=mRNA0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP_**002296275.1,GeneID:7008893;** gbkey=CDS;product=hypothetical protein;protein_id=YP_**002296275.1;transl_table=11
I re-ran cufflink however this time there is error reported. I can only tell from the report that there is a segmentation fault but not further details. The report is as follows:
Error running cufflinks. return code = 139 Command line: cufflinks -q --no-update-check -I 100 -F 0.100000 -j 0.150000 -p 4 -G /galaxy/test_pool/pool5/files/**000/327/dataset_327777.dat /galaxy/test_database/files/**000/325/dataset_325086.dat [19:41:41] Loading reference annotation. Segmentation fault
cp: cannot stat `/galaxy/test_pool/pool3/tmp/** job_working_directory/000/170/**170197/global_model.txt': No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/** job_working_directory/000/170/**170197/isoforms.fpkm_tracking'**: No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/** job_working_directory/000/170/**170197/genes.fpkm_tracking': No such file or directory
My questions will be:
1. Is there any way to modify a NCBI bacterial genome annotation GFF file to make it usable for cufflink? Our genome annotation is only available in NCBI, not ensemble or USDC so this is pretty much my only choice..
2. Should I proceed with modifying the GFF file or should I convert it into GTF and use the GTF instead in cufflink?
I am a biochemist and really new to the computer world so any advice will help!
Thanks a lot,
Qian -- Qian Dong Bauer Lab, MCBD Simon Hall: 313-317 212 S. Hawthorne Dr. Bloomington, IN 47405 Email:dong3@indiana.edu <mailto:Email%3Adong3@indiana.**edu<Email%253Adong3@indiana.edu>
Lab Phone:812-855-8443
______________________________**_____________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/**listinfo/galaxy-dev<http://lists.bx.psu.edu/listinfo/galaxy-dev>
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://galaxyproject.org
-- Qian Dong Bauer Lab, MCBD Simon Hall: 313-317 212 S. Hawthorne Dr. Bloomington, IN 47405 Email:dong3@indiana.edu Lab Phone:812-855-8443
Hi Qian, This is the source of the mismatch. In fasta format, any text between the leading ">" and the first whitespace (tab, space, etc) is considered the "identifier". http://wiki.g2.bx.psu.edu/Support#Error_from_tools http://wiki.g2.bx.psu.edu/Learn/Datatypes#Fasta Which in your case is this: gi|289546492|ref|NC_011420.2| The identifier must be changed to be only this: NC_011420.2 to be consistent with the GFF file. There are many ways to do this, one simple way is to use the tools in 'Text Manipulation'. Use 'Remove beginning of a file' to strip the first line. Then create a plain text file on your computer, one line, with just the identifier content like this: >NC_011420.2 Very important - do not make the file more than one line long - meaning do not add in an extra 'return' at the end of the first line. The plain text file just needs these characters - no more - including no whitespace characters (spaces, tabs, newlines). Upload this file to your instance as text and "Concatenate datasets" with the sequence file that had the first line removed, placing this identifier dataset on top. Then change the datatype back to ".fasta" on the final dataset, if necessary (click on the pencil icon). Then re-run Bowtie to have the identifiers in your SAM file consistent with the GFF file. After running Bowtie, be certain to sort the resulting dataset before running Cufflinks. Tophat produces sorted output, but Bowtie does not and Cufflinks requires sorted input. http://main.g2.bx.psu.edu/u/jeremy/p/transcriptome-analysis-faq#faq2 Take care, Jen Galaxy team On 9/26/12 12:54 PM, Qian Dong wrote:
Dear Jennifer,
My .fasta reference genome is like this:
'>gi|289546492|ref|NC_011420.2| Rhodospirillum centenum SW chromosome, complete genome'
and in the SAM file generated by BOWTIE it says:
@SQ SN:gi|289546492|ref|NC_011420.2| LN:4355543
So I think they are all the same as "NC_011420.2". Is there anything else I can try?
Thank you,
Qian
On Wed, Sep 26, 2012 at 3:35 PM, Jennifer Jackson <jen@bx.psu.edu <mailto:jen@bx.psu.edu>> wrote:
Hello,
The first thing to double check is that the chromosome identifier is an exact match between the reference genome and the reference annotation.
The GFF3 file is naming the chromosome "NC_011420.2".
The reference annotation chromosome should be named exactly the same way. Check this in the input BAM/SAM datasets or the original .fasta reference genome.
Hopefully this finds the problem. Correcting mismatched names (due to various reasons) is the most common solution to this sort of issue: 'Tools on the Main server: Example', bullet item #2: http://wiki.g2.bx.psu.edu/__Support#Interpreting___scientific_results <http://wiki.g2.bx.psu.edu/Support#Interpreting_scientific_results>
Best,
Jen Galaxy team
On 9/26/12 8:46 AM, Qian Dong wrote:
Dear Team,
I've been having a problem with cufflink regarding GFF files. I tried searching the mailing list first and failed to find an answer. Could you help me look at this?
I downloaded my genome annotation GFF file from NCBI (soon I realized NCBI format may be a problem) for my bacterial RNA-seq data analysis. My GFF file looks like the following:
' ##gff-version 3 #!gff-spec-version 1.20 #!processor NCBI annotwriter ##sequence-region NC_011420.2 1 4355543 <tel:011420.2%201%204355543> ##species http://www.ncbi.nlm.nih.gov/__Taxonomy/Browser/wwwtax.cgi?__id=414684 <http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684> NC_011420.2 RefSeq region 1 4355543 . + . ID=id0;Dbxref=taxon:414684;Is___circular=true;culture-__collection=ATCC:51521;gb-__synonym=Rhodocista centenaria SW;gbkey=Src;genome=__chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521 NC_011420.2 RefSeq gene 11 3343 . + . ID=gene0;Name=RC1_0011;Dbxref=__GeneID:7008893;gbkey=Gene;__locus_tag=RC1_0011 NC_011420.2 RefSeq CDS 11 3343 . + 0 ID=cds0;Name=YP_002296275.1;__Parent=gene0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP___002296275.1,GeneID:7008893;__gbkey=CDS;product=hypothetical protein;protein_id=YP___002296275.1;transl_table=11
I used this file for cufflink but all the FPKM values are 0. I checked out this link: http://cufflinks.cbcb.umd.edu/__gff.html <http://cufflinks.cbcb.umd.edu/gff.html> and thought that maybe the problem is because I don't have any mRNA feature in my gff file. Since I am dealing with a bacterial genome, there is no exon/intron or UTR info needed. Therefore I modified my GFF file into the following:
##gff-version 3 #!gff-spec-version 1.20 #!processor NCBI annotwriter ##sequence-region NC_011420.2 1 4355543 <tel:011420.2%201%204355543> ##species http://www.ncbi.nlm.nih.gov/__Taxonomy/Browser/wwwtax.cgi?__id=414684 <http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=414684> NC_011420.2 RefSeq region 1 4355543 . + . ID=id0;Dbxref=taxon:414684;Is___circular=true;culture-__collection=ATCC:51521;gb-__synonym=Rhodocista centenaria SW;gbkey=Src;genome=__chromosome;mol_type=genomic DNA;strain=SW%3B ATCC 51521 NC_011420.2 RefSeq mRNA 11 3343 . + . ID=mRNA0;Name=RC1_0011;Dbxref=__GeneID:7008893;gbkey=Gene;__locus_tag=RC1_0011 NC_011420.2 RefSeq CDS 11 3343 . + 0 ID=cds0;Name=YP_002296275.1;__Parent=mRNA0;Note=Contains a type I secretion target ggxgxdxxx repeat %282 copies%29 domain%3B Contains a Cadherin domain%3B identified by match to protein family HMM PF02789;Dbxref=Genbank:YP___002296275.1,GeneID:7008893;__gbkey=CDS;product=hypothetical protein;protein_id=YP___002296275.1;transl_table=11
I re-ran cufflink however this time there is error reported. I can only tell from the report that there is a segmentation fault but not further details. The report is as follows:
Error running cufflinks. return code = 139 Command line: cufflinks -q --no-update-check -I 100 -F 0.100000 -j 0.150000 -p 4 -G /galaxy/test_pool/pool5/files/__000/327/dataset_327777.dat /galaxy/test_database/files/__000/325/dataset_325086.dat [19:41:41] Loading reference annotation. Segmentation fault
cp: cannot stat `/galaxy/test_pool/pool3/tmp/__job_working_directory/000/170/__170197/global_model.txt': No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/__job_working_directory/000/170/__170197/isoforms.fpkm_tracking'__: No such file or directory cp: cannot stat `/galaxy/test_pool/pool3/tmp/__job_working_directory/000/170/__170197/genes.fpkm_tracking': No such file or directory
My questions will be:
1. Is there any way to modify a NCBI bacterial genome annotation GFF file to make it usable for cufflink? Our genome annotation is only available in NCBI, not ensemble or USDC so this is pretty much my only choice..
2. Should I proceed with modifying the GFF file or should I convert it into GTF and use the GTF instead in cufflink?
I am a biochemist and really new to the computer world so any advice will help!
Thanks a lot,
Qian -- Qian Dong Bauer Lab, MCBD Simon Hall: 313-317 212 S. Hawthorne Dr. Bloomington, IN 47405 Email:dong3@indiana.edu <mailto:Email%3Adong3@indiana.edu> <mailto:Email%3Adong3@indiana.__edu <mailto:Email%253Adong3@indiana.edu>> Lab Phone:812-855-8443 <tel:812-855-8443>
_____________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org <http://usegalaxy.org>. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/__listinfo/galaxy-dev <http://lists.bx.psu.edu/listinfo/galaxy-dev>
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Jennifer Jackson http://galaxyproject.org
-- Qian Dong Bauer Lab, MCBD Simon Hall: 313-317 212 S. Hawthorne Dr. Bloomington, IN 47405 Email:dong3@indiana.edu <mailto:Email%3Adong3@indiana.edu> Lab Phone:812-855-8443
-- Jennifer Jackson http://galaxyproject.org
participants (3)
-
 Jennifer Jackson
Jennifer Jackson -
 Kenneth Auerbach
Kenneth Auerbach -
 Qian Dong
Qian Dong