Normalization an dplotting of RPKM/FPKM after cufflink

Hi, I want to include the following discussion in my message regarding use Bam files of Tophat to visualize reads either in IGV or Galaxy or other tools. I want to find out if I can plot RPKM/FPKM normalized values after running differential analysis in Cufflinks. On the seqanswer (http://seqanswers.com/forums/showthread.php?t=9947 ) there is a preliminary discussion about this how we can plot RPKM values to show the differential abundance in samples. Do we have any of such functionality in Galaxy? alternatively, I would like to have suggestions on the topic especially to normalize per million reads. Thanks. --- On Tue, 2/22/11, Jeremy Goecks <jeremy.goecks@emory.edu> wrote: From: Jeremy Goecks <jeremy.goecks@emory.edu> Subject: Re: [galaxy-user] get wig file after tophat To: "David Matthews" <D.A.Matthews@bristol.ac.uk> Cc: "Baxter, Adam" <Adam.Baxter@uncc.edu>, galaxy-user@bx.psu.edu Date: Tuesday, February 22, 2011, 11:30 AM All, For visualization, Galaxy now provides a built-in browser called Trackster. Trackster can visualize BAM files--as well as the junction file produced by Tophat and the GFF files produced by Cufflinks--and also provides coverage/summary information for all datatypes (see attached image). You can start using Trackster by going to the Visualization --> New Track Browser to set up a browser and add tracks. Alternatively, you can click on the Trackster icon in a dataset to visualize the dataset in a new or existing visualization (see attached image). Thanks, J. On Feb 22, 2011, at 11:54 AM, David Matthews wrote: HI, The option you need in IGV tools is "count". You set a window size and this gives you a tdf file from your sorted bam (or sam) file which is nice and quick to view on IGV. Best Wishes, David. __________________________________ Dr David A. Matthews Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K. Tel. +44 117 3312058 Fax. +44 117 3312091 D.A.Matthews@bristol.ac.uk On 22 Feb 2011, at 15:52, Ying Zhang wrote: Dear David: thank you very much for helping me! I have download the IGV and I do find the IGVtools, however, I am not sure which tool I should use for generate a tdf file, the tile function will generate a tdf file, but the input file format does not include bam or sam file, instead it need wig file. But I have no wig file to put in. So I am wondering whether you need to use other tool first. I really appreciate your help! Thank you very much! Best Ying Quoting David Matthews <D.A.Matthews@bristol.ac.uk>: Hi, You can get an equivalent visualisation from the IGV viewer by the Broad Institute - its under IGV tools and generates a tdf file from bam or sam files. This also gives a quick and easy way of looking at depth at any particular site and is very accessible. Cheers David On 21 Feb 2011, at 21:44, Jeremy Goecks wrote: Hi all, Ann is correct - Tophat does not produce .wig files when run anymore. However, it's fairly easy to use Galaxy to make a wiggle-like coverage file from a BAM file: (a) run the pileup tool on your BAM to create a pileup file; (b) cut columns 1 and 4 to get your coverage file. A final note: it's often difficult to visualize coverage files because they're so large. You might be better off visualizing the BAM file and using the coverage file for statistics. Best, J. Hello, I think I know the answer (sort of) to this question. This may be because newer versions of tophat stopped running the "wiggles" program, which is still part of the tophat distribution and is the program that makes the "coverage.wig" file. A later version of tophat might bring this back, however - there's a note to this effect in the tophat python code. So if you can run wiggles, you can make the "coverage.wig" file on your own. A student here at UNC Charlotte (Adam Baxter) made a few changes to the "wiggles" source code that would allow you to use it with samtools to make a "coverage.wig" file from the "accepted_hits.bam" file that TopHat creates. If you (or anyone else) would like a copy, please email Adam, who is cc'ed on this email. We would be happy to help add it to Galaxy if this would be of interest to you or other Galaxy users. If there is any way we can be of assistance, please let us know! Very best wishes, Ann Loraine On 2/21/11 3:39 PM, "Ying Zhang" <ying.zhang.yz323@yale.edu> wrote: Hi: I am using tophat in galaxy to analyze my paired-end RNA-seq data and find out that after the tophat analysis, we can not get the wig file from it anymore which is used to be able to. Do you have any idea of how to still be able to get the wig file after tophat analysis? Thanks a lot! Best Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286 _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ -- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286 -----Inline Attachment Follows----- _______________________________________________ /

Hi, This is not a Galaxy answer specifically, but since you mention IGV, you can load the "expr" and of course "gtf" files in IGV 2.0. Its not released but is available from the "early access" links on the downloads page (htttp://www.broadinstitute.org/igv/downloads). If you want to use IGV with Galaxy we have some XML that will enable that, email me at igv-help@broadinstitute.org to request it. Best, Jim
Hi,
I want to include the following discussion in my message regarding use Bam files of Tophat to visualize reads either in IGV or Galaxy or other tools. I want to find out if I can plot RPKM/FPKM normalized values after running differential analysis in Cufflinks. On the seqanswer (http://seqanswers.com/forums/showthread.php?t=9947 ) there is a preliminary discussion about this how we can plot RPKM values to show the differential abundance in samples. Do we have any of such functionality in Galaxy? alternatively, I would like to have suggestions on the topic especially to normalize per million reads.
Thanks.
--- On Tue, 2/22/11, Jeremy Goecks <jeremy.goecks@emory.edu> wrote:
From: Jeremy Goecks <jeremy.goecks@emory.edu> Subject: Re: [galaxy-user] get wig file after tophat To: "David Matthews" <D.A.Matthews@bristol.ac.uk> Cc: "Baxter, Adam" <Adam.Baxter@uncc.edu>, galaxy-user@bx.psu.edu Date: Tuesday, February 22, 2011, 11:30 AM
All,
For visualization, Galaxy now provides a built-in browser called Trackster. Trackster can visualize BAM files--as well as the junction file produced by Tophat and the GFF files produced by Cufflinks--and also provides coverage/summary information for all datatypes (see attached image).
You can start using Trackster by going to the Visualization --> New Track Browser to set up a browser and add tracks. Alternatively, you can click on the Trackster icon in a dataset to visualize the dataset in a new or existing visualization (see attached image).
Thanks, J.
On Feb 22, 2011, at 11:54 AM, David Matthews wrote:
HI,
The option you need in IGV tools is "count". You set a window size and this gives you a tdf file from your sorted bam (or sam) file which is nice and quick to view on IGV.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
On 22 Feb 2011, at 15:52, Ying Zhang wrote:
Dear David:
thank you very much for helping me!
I have download the IGV and I do find the IGVtools, however, I am not sure which tool I should use for generate a tdf file, the tile function will generate a tdf file, but the input file format does not include bam or sam file, instead it need wig file. But I have no wig file to put in. So I am wondering whether you need to use other tool first. I really appreciate your help! Thank you very much!
Best
Ying
Quoting David Matthews <D.A.Matthews@bristol.ac.uk>:
Hi,
You can get an equivalent visualisation from the IGV viewer by the Broad Institute - its under IGV tools and generates a tdf file from bam or sam files. This also gives a quick and easy way of looking at depth at any particular site and is very accessible.
Cheers David
On 21 Feb 2011, at 21:44, Jeremy Goecks wrote:
Hi all,
Ann is correct - Tophat does not produce .wig files when run anymore. However, it's fairly easy to use Galaxy to make a wiggle-like coverage file from a BAM file:
(a) run the pileup tool on your BAM to create a pileup file; (b) cut columns 1 and 4 to get your coverage file.
A final note: it's often difficult to visualize coverage files because they're so large. You might be better off visualizing the BAM file and using the coverage file for statistics.
Best, J.
Hello,
I think I know the answer (sort of) to this question.
This may be because newer versions of tophat stopped running the "wiggles" program, which is still part of the tophat distribution and is the program that makes the "coverage.wig" file.
A later version of tophat might bring this back, however - there's a note to this effect in the tophat python code.
So if you can run wiggles, you can make the "coverage.wig" file on your own.
A student here at UNC Charlotte (Adam Baxter) made a few changes to the "wiggles" source code that would allow you to use it with samtools to make a "coverage.wig" file from the "accepted_hits.bam" file that TopHat creates.
If you (or anyone else) would like a copy, please email Adam, who is cc'ed on this email.
We would be happy to help add it to Galaxy if this would be of interest to you or other Galaxy users.
If there is any way we can be of assistance, please let us know!
Very best wishes,
Ann Loraine
On 2/21/11 3:39 PM, "Ying Zhang" <ying.zhang.yz323@yale.edu> wrote:
> Hi: > > I am using tophat in galaxy to analyze my paired-end RNA-seq > data and find out > that after the tophat analysis, we can not get the wig file > from it anymore > which is used to be able to. Do you have any idea of how to > still be able to > get the wig file after tophat analysis? Thanks a lot! > > Best > > Ying Zhang, M.D., Ph.D. > Postdoctoral Associate > Department of Genetics, > Yale University School of Medicine > 300 Cedar Street,S320 > New Haven, CT 06519 > Tel: (203)737-2616 > Fax: (203)737-2286 > _______________________________________________ > The Galaxy User list should be used for the discussion > of Galaxy analysis and other features on the public > server at usegalaxy.org. For discussion of local Galaxy > instances and the Galaxy source code, please use the > Galaxy Development list: > > http://lists.bx.psu.edu/listinfo/galaxy-dev > > To manage your subscriptions to this and other > Galaxy lists, please use the interface at: > > http://lists.bx.psu.edu/
-- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286
-----Inline Attachment Follows-----
_______________________________________________ / ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:

Hello, We would like to mention that a community contributed XML configuration file that enables BAM display at IGV was added to Galaxy in 5252:a62cec23a157. In your local Galaxy instance, you can enable it by removing the comments from around: <!-- <display file="igv/bam.xml" /> --> in your datatypes_conf.xml file for the BAM datatype. This will let users view their BAM files in a local IGV instance or using web start. We would be interested in including additional displays in Galaxy for other datatypes when they are supported in IGV. If configurations already exist for other datatypes, please feel free to contribute them. Thanks for using Galaxy, Dan On Apr 14, 2011, at 7:56 AM, Jim Robinson wrote:
Hi,
This is not a Galaxy answer specifically, but since you mention IGV, you can load the "expr" and of course "gtf" files in IGV 2.0. Its not released but is available from the "early access" links on the downloads page (htttp://www.broadinstitute.org/igv/downloads). If you want to use IGV with Galaxy we have some XML that will enable that, email me at igv-help@broadinstitute.org to request it.
Best,
Jim
Hi,
I want to include the following discussion in my message regarding use Bam files of Tophat to visualize reads either in IGV or Galaxy or other tools. I want to find out if I can plot RPKM/FPKM normalized values after running differential analysis in Cufflinks. On the seqanswer (http://seqanswers.com/forums/showthread.php?t=9947 ) there is a preliminary discussion about this how we can plot RPKM values to show the differential abundance in samples. Do we have any of such functionality in Galaxy? alternatively, I would like to have suggestions on the topic especially to normalize per million reads.
Thanks.
--- On Tue, 2/22/11, Jeremy Goecks <jeremy.goecks@emory.edu> wrote:
From: Jeremy Goecks <jeremy.goecks@emory.edu> Subject: Re: [galaxy-user] get wig file after tophat To: "David Matthews" <D.A.Matthews@bristol.ac.uk> Cc: "Baxter, Adam" <Adam.Baxter@uncc.edu>, galaxy-user@bx.psu.edu Date: Tuesday, February 22, 2011, 11:30 AM
All,
For visualization, Galaxy now provides a built-in browser called Trackster. Trackster can visualize BAM files--as well as the junction file produced by Tophat and the GFF files produced by Cufflinks--and also provides coverage/summary information for all datatypes (see attached image).
You can start using Trackster by going to the Visualization --> New Track Browser to set up a browser and add tracks. Alternatively, you can click on the Trackster icon in a dataset to visualize the dataset in a new or existing visualization (see attached image).
Thanks, J.
On Feb 22, 2011, at 11:54 AM, David Matthews wrote:
HI,
The option you need in IGV tools is "count". You set a window size and this gives you a tdf file from your sorted bam (or sam) file which is nice and quick to view on IGV.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
On 22 Feb 2011, at 15:52, Ying Zhang wrote:
Dear David:
thank you very much for helping me!
I have download the IGV and I do find the IGVtools, however, I am not sure which tool I should use for generate a tdf file, the tile function will generate a tdf file, but the input file format does not include bam or sam file, instead it need wig file. But I have no wig file to put in. So I am wondering whether you need to use other tool first. I really appreciate your help! Thank you very much!
Best
Ying
Quoting David Matthews <D.A.Matthews@bristol.ac.uk>:
Hi,
You can get an equivalent visualisation from the IGV viewer by the Broad Institute - its under IGV tools and generates a tdf file from bam or sam files. This also gives a quick and easy way of looking at depth at any particular site and is very accessible.
Cheers David
On 21 Feb 2011, at 21:44, Jeremy Goecks wrote:
Hi all,
Ann is correct - Tophat does not produce .wig files when run anymore. However, it's fairly easy to use Galaxy to make a wiggle-like coverage file from a BAM file:
(a) run the pileup tool on your BAM to create a pileup file; (b) cut columns 1 and 4 to get your coverage file.
A final note: it's often difficult to visualize coverage files because they're so large. You might be better off visualizing the BAM file and using the coverage file for statistics.
Best, J.
> Hello, > > I think I know the answer (sort of) to this question. > > This may be because newer versions of tophat stopped running the "wiggles" > program, which is still part of the tophat distribution and is the program > that makes the "coverage.wig" file. > > A later version of tophat might bring this back, however - there's a note to > this effect in the tophat python code. > > So if you can run wiggles, you can make the "coverage.wig" file on your own. > > A student here at UNC Charlotte (Adam Baxter) made a few changes to the > "wiggles" source code that would allow you to use it with samtools to make a > "coverage.wig" file from the "accepted_hits.bam" file that TopHat creates. > > If you (or anyone else) would like a copy, please email Adam, who is cc'ed > on this email. > > We would be happy to help add it to Galaxy if this would be of interest to > you or other Galaxy users. > > If there is any way we can be of assistance, please let us know! > > Very best wishes, > > Ann Loraine > > > On 2/21/11 3:39 PM, "Ying Zhang" <ying.zhang.yz323@yale.edu> wrote: > >> Hi: >> >> I am using tophat in galaxy to analyze my paired-end RNA-seq data and find out >> that after the tophat analysis, we can not get the wig file from it anymore >> which is used to be able to. Do you have any idea of how to still be able to >> get the wig file after tophat analysis? Thanks a lot! >> >> Best >> >> Ying Zhang, M.D., Ph.D. >> Postdoctoral Associate >> Department of Genetics, >> Yale University School of Medicine >> 300 Cedar Street,S320 >> New Haven, CT 06519 >> Tel: (203)737-2616 >> Fax: (203)737-2286 >> _______________________________________________ >> The Galaxy User list should be used for the discussion >> of Galaxy analysis and other features on the public >> server at usegalaxy.org. For discussion of local Galaxy >> instances and the Galaxy source code, please use the >> Galaxy Development list: >> >> http://lists.bx.psu.edu/listinfo/galaxy-dev >> >> To manage your subscriptions to this and other >> Galaxy lists, please use the interface at: >> >> http://lists.bx.psu.edu/ > > -- > Ann Loraine > Associate Professor > Dept. of Bioinformatics and Genomics, UNCC > North Carolina Research Campus > 600 Laureate Way > Kannapolis, NC 28081 > 704-250-5750 > www.transvar.org > > > _______________________________________________ > The Galaxy User list should be used for the discussion > of Galaxy analysis and other features on the public > server at usegalaxy.org. For discussion of local Galaxy > instances and the Galaxy source code, please use the > Galaxy Development list: > > http://lists.bx.psu.edu/listinfo/galaxy-dev > > To manage your subscriptions to this and other > Galaxy lists, please use the interface at: > > http://lists.bx.psu.edu/
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286
-----Inline Attachment Follows-----
_______________________________________________ / ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Jim, Thanks. My idea is to explore any option for visualizing RPKM in IGV or Galaxy or any other common tool. Since Galaxy has created some additional visualization track with Genome browser so I want to have feedback from other users. Thanks --- On Thu, 4/14/11, Daniel Blankenberg <dan@bx.psu.edu> wrote: From: Daniel Blankenberg <dan@bx.psu.edu> Subject: Re: [galaxy-user] Normalization an dplotting of RPKM/FPKM after cufflink To: "Jim Robinson" <jrobinso@broadinstitute.org> Cc: "vasu punj" <punjv@yahoo.com>, galaxy-user@bx.psu.edu Date: Thursday, April 14, 2011, 7:49 AM Hello, We would like to mention that a community contributed XML configuration file that enables BAM display at IGV was added to Galaxy in 5252:a62cec23a157. In your local Galaxy instance, you can enable it by removing the comments from around: <!-- <display file="igv/bam.xml" /> --> in your datatypes_conf.xml file for the BAM datatype. This will let users view their BAM files in a local IGV instance or using web start. We would be interested in including additional displays in Galaxy for other datatypes when they are supported in IGV. If configurations already exist for other datatypes, please feel free to contribute them. Thanks for using Galaxy, Dan On Apr 14, 2011, at 7:56 AM, Jim Robinson wrote:
Hi,
This is not a Galaxy answer specifically, but since you mention IGV, you can load the "expr" and of course "gtf" files in IGV 2.0. Its not released but is available from the "early access" links on the downloads page (htttp://www.broadinstitute.org/igv/downloads). If you want to use IGV with Galaxy we have some XML that will enable that, email me at igv-help@broadinstitute.org to request it.
Best,
Jim
Hi,
I want to include the following discussion in my message regarding use Bam files of Tophat to visualize reads either in IGV or Galaxy or other tools. I want to find out if I can plot RPKM/FPKM normalized values after running differential analysis in Cufflinks. On the seqanswer (http://seqanswers.com/forums/showthread.php?t=9947 ) there is a preliminary discussion about this how we can plot RPKM values to show the differential abundance in samples. Do we have any of such functionality in Galaxy? alternatively, I would like to have suggestions on the topic especially to normalize per million reads.
Thanks.
--- On Tue, 2/22/11, Jeremy Goecks <jeremy.goecks@emory.edu> wrote:
From: Jeremy Goecks <jeremy.goecks@emory.edu> Subject: Re: [galaxy-user] get wig file after tophat To: "David Matthews" <D.A.Matthews@bristol.ac.uk> Cc: "Baxter, Adam" <Adam.Baxter@uncc.edu>, galaxy-user@bx.psu.edu Date: Tuesday, February 22, 2011, 11:30 AM
All,
For visualization, Galaxy now provides a built-in browser called Trackster. Trackster can visualize BAM files--as well as the junction file produced by Tophat and the GFF files produced by Cufflinks--and also provides coverage/summary information for all datatypes (see attached image).
You can start using Trackster by going to the Visualization --> New Track Browser to set up a browser and add tracks. Alternatively, you can click on the Trackster icon in a dataset to visualize the dataset in a new or existing visualization (see attached image).
Thanks, J.
On Feb 22, 2011, at 11:54 AM, David Matthews wrote:
HI,
The option you need in IGV tools is "count". You set a window size and this gives you a tdf file from your sorted bam (or sam) file which is nice and quick to view on IGV.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
On 22 Feb 2011, at 15:52, Ying Zhang wrote:
Dear David:
thank you very much for helping me!
I have download the IGV and I do find the IGVtools, however, I am not sure which tool I should use for generate a tdf file, the tile function will generate a tdf file, but the input file format does not include bam or sam file, instead it need wig file. But I have no wig file to put in. So I am wondering whether you need to use other tool first. I really appreciate your help! Thank you very much!
Best
Ying
Quoting David Matthews <D.A.Matthews@bristol.ac.uk>:
Hi,
You can get an equivalent visualisation from the IGV viewer by the Broad Institute - its under IGV tools and generates a tdf file from bam or sam files. This also gives a quick and easy way of looking at depth at any particular site and is very accessible.
Cheers David
On 21 Feb 2011, at 21:44, Jeremy Goecks wrote:
Hi all,
Ann is correct - Tophat does not produce .wig files when run anymore. However, it's fairly easy to use Galaxy to make a wiggle-like coverage file from a BAM file:
(a) run the pileup tool on your BAM to create a pileup file; (b) cut columns 1 and 4 to get your coverage file.
A final note: it's often difficult to visualize coverage files because they're so large. You might be better off visualizing the BAM file and using the coverage file for statistics.
Best, J.
> Hello, > > I think I know the answer (sort of) to this question. > > This may be because newer versions of tophat stopped running the "wiggles" > program, which is still part of the tophat distribution and is the program > that makes the "coverage.wig" file. > > A later version of tophat might bring this back, however - there's a note to > this effect in the tophat python code. > > So if you can run wiggles, you can make the "coverage.wig" file on your own. > > A student here at UNC Charlotte (Adam Baxter) made a few changes to the > "wiggles" source code that would allow you to use it with samtools to make a > "coverage.wig" file from the "accepted_hits.bam" file that TopHat creates. > > If you (or anyone else) would like a copy, please email Adam, who is cc'ed > on this email. > > We would be happy to help add it to Galaxy if this would be of interest to > you or other Galaxy users. > > If there is any way we can be of assistance, please let us know! > > Very best wishes, > > Ann Loraine > > > On 2/21/11 3:39 PM, "Ying Zhang" <ying.zhang.yz323@yale.edu> wrote: > >> Hi: >> >> I am using tophat in galaxy to analyze my paired-end RNA-seq data and find out >> that after the tophat analysis, we can not get the wig file from it anymore >> which is used to be able to. Do you have any idea of how to still be able to >> get the wig file after tophat analysis? Thanks a lot! >> >> Best >> >> Ying Zhang, M.D., Ph.D. >> Postdoctoral Associate >> Department of Genetics, >> Yale University School of Medicine >> 300 Cedar Street,S320 >> New Haven, CT 06519 >> Tel: (203)737-2616 >> Fax: (203)737-2286 >> _______________________________________________ >> The Galaxy User list should be used for the discussion >> of Galaxy analysis and other features on the public >> server at usegalaxy.org. For discussion of local Galaxy >> instances and the Galaxy source code, please use the >> Galaxy Development list: >> >> http://lists.bx.psu.edu/listinfo/galaxy-dev >> >> To manage your subscriptions to this and other >> Galaxy lists, please use the interface at: >> >> http://lists.bx.psu.edu/ > > -- > Ann Loraine > Associate Professor > Dept. of Bioinformatics and Genomics, UNCC > North Carolina Research Campus > 600 Laureate Way > Kannapolis, NC 28081 > 704-250-5750 > www.transvar.org > > > _______________________________________________ > The Galaxy User list should be used for the discussion > of Galaxy analysis and other features on the public > server at usegalaxy.org. For discussion of local Galaxy > instances and the Galaxy source code, please use the > Galaxy Development list: > > http://lists.bx.psu.edu/listinfo/galaxy-dev > > To manage your subscriptions to this and other > Galaxy lists, please use the interface at: > > http://lists.bx.psu.edu/
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286
-----Inline Attachment Follows-----
_______________________________________________ / ___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
___________________________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. Please keep all replies on the list by using "reply all" in your mail client. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:

Vasu, It's not clear what you're trying to accomplish, but I'll try to provide some information that you might find useful:
I want to include the following discussion in my message regarding use Bam files of Tophat to visualize reads either in IGV or Galaxy or other tools. I want to find out if I can plot RPKM/FPKM normalized values after running differential analysis in Cufflinks.
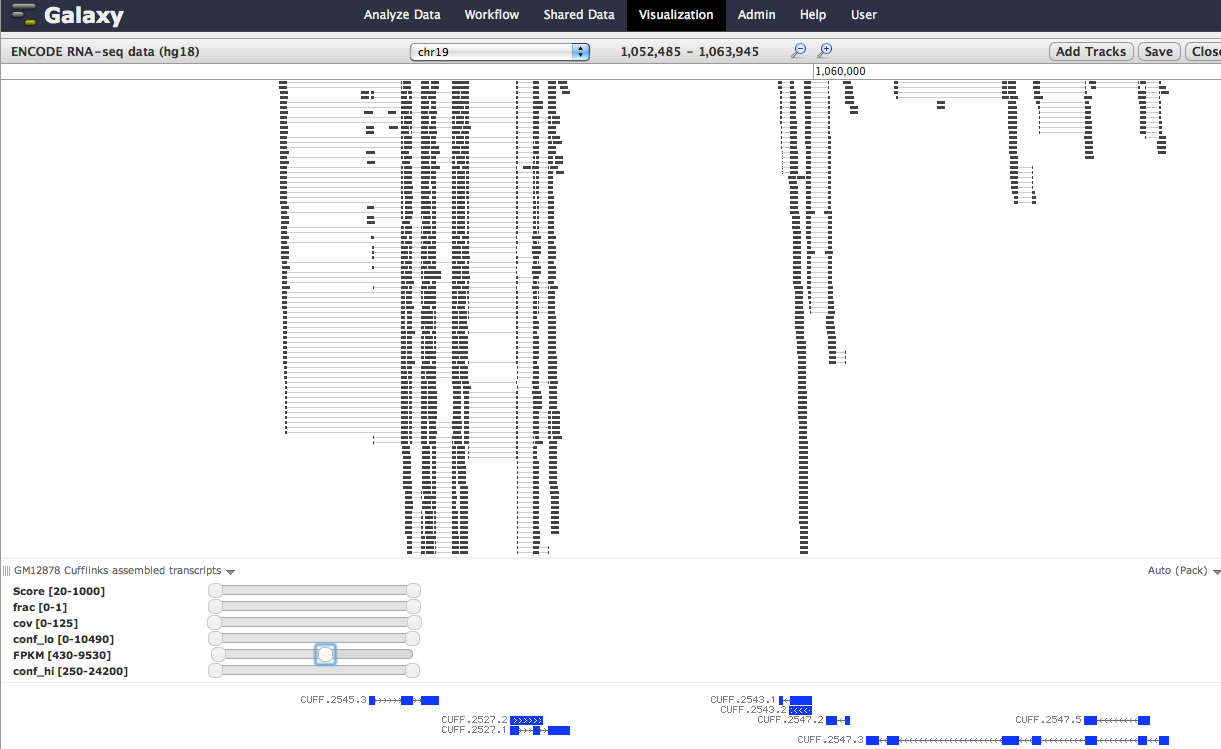
Galaxy has a number of tools for analyzing numerical data; look under the menu items Statistics and Graph/Display Data for useful tools. If you're looking to plot FPKM values in addition to mapped reads from Tophat and Cufflinks transcripts, the Galaxy Tracks Browser might prove useful as it has filtering functionality so that you can move a slider to show/hide data based on FPKM values; its often useful to use the sliders for FPKM measures to get a sense of your data. See the attached screenshot for an example.
On the seqanswer (http://seqanswers.com/forums/showthread.php?t=9947 ) there is a preliminary discussion about this how we can plot RPKM values to show the differential abundance in samples. Do we have any of such functionality in Galaxy? alternatively, I would like to have suggestions on the topic especially to normalize per million reads.
This thread discusses alternative approaches for computing RPKM/FPKM from mapped read data. If you choose not to use Cufflinks/compare/diff to compute RPKM, you can compute a rough RPKM by doing the following: (1) use the pileup tool to compute coverage across a genome; (2) divide coverage by the total number of mapped reads (in millions) to get coverage per million reads (CPM); and (3) average CPM across a transcript and divide by the length of the transcript to get ~RPKM. Best, J.
{kind=link}
participants (4)
-
 Daniel Blankenberg
Daniel Blankenberg -
 Jeremy Goecks
Jeremy Goecks -
 Jim Robinson
Jim Robinson -
 vasu punj
vasu punj