Re: [galaxy-user] get wig file after tophat

HI, The option you need in IGV tools is "count". You set a window size and this gives you a tdf file from your sorted bam (or sam) file which is nice and quick to view on IGV. Best Wishes, David. __________________________________ Dr David A. Matthews Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K. Tel. +44 117 3312058 Fax. +44 117 3312091 D.A.Matthews@bristol.ac.uk On 22 Feb 2011, at 15:52, Ying Zhang wrote:
Dear David:
thank you very much for helping me!
I have download the IGV and I do find the IGVtools, however, I am not sure which tool I should use for generate a tdf file, the tile function will generate a tdf file, but the input file format does not include bam or sam file, instead it need wig file. But I have no wig file to put in. So I am wondering whether you need to use other tool first. I really appreciate your help! Thank you very much!
Best
Ying
Quoting David Matthews <D.A.Matthews@bristol.ac.uk>:
Hi,
You can get an equivalent visualisation from the IGV viewer by the Broad Institute - its under IGV tools and generates a tdf file from bam or sam files. This also gives a quick and easy way of looking at depth at any particular site and is very accessible.
Cheers David
On 21 Feb 2011, at 21:44, Jeremy Goecks wrote:
Hi all,
Ann is correct - Tophat does not produce .wig files when run anymore. However, it's fairly easy to use Galaxy to make a wiggle-like coverage file from a BAM file:
(a) run the pileup tool on your BAM to create a pileup file; (b) cut columns 1 and 4 to get your coverage file.
A final note: it's often difficult to visualize coverage files because they're so large. You might be better off visualizing the BAM file and using the coverage file for statistics.
Best, J.
Hello,
I think I know the answer (sort of) to this question.
This may be because newer versions of tophat stopped running the "wiggles" program, which is still part of the tophat distribution and is the program that makes the "coverage.wig" file.
A later version of tophat might bring this back, however - there's a note to this effect in the tophat python code.
So if you can run wiggles, you can make the "coverage.wig" file on your own.
A student here at UNC Charlotte (Adam Baxter) made a few changes to the "wiggles" source code that would allow you to use it with samtools to make a "coverage.wig" file from the "accepted_hits.bam" file that TopHat creates.
If you (or anyone else) would like a copy, please email Adam, who is cc'ed on this email.
We would be happy to help add it to Galaxy if this would be of interest to you or other Galaxy users.
If there is any way we can be of assistance, please let us know!
Very best wishes,
Ann Loraine
On 2/21/11 3:39 PM, "Ying Zhang" <ying.zhang.yz323@yale.edu> wrote:
Hi:
I am using tophat in galaxy to analyze my paired-end RNA-seq data and find out that after the tophat analysis, we can not get the wig file from it anymore which is used to be able to. Do you have any idea of how to still be able to get the wig file after tophat analysis? Thanks a lot!
Best
Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286 _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286

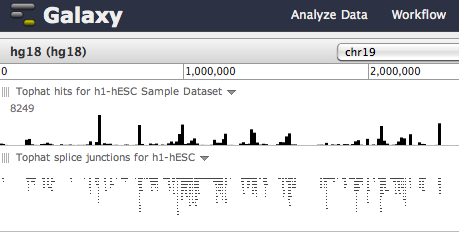
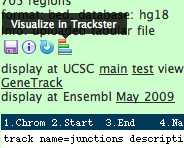
All, For visualization, Galaxy now provides a built-in browser called Trackster. Trackster can visualize BAM files--as well as the junction file produced by Tophat and the GFF files produced by Cufflinks--and also provides coverage/summary information for all datatypes (see attached image). You can start using Trackster by going to the Visualization --> New Track Browser to set up a browser and add tracks. Alternatively, you can click on the Trackster icon in a dataset to visualize the dataset in a new or existing visualization (see attached image). Thanks, J. On Feb 22, 2011, at 11:54 AM, David Matthews wrote:
HI,
The option you need in IGV tools is "count". You set a window size and this gives you a tdf file from your sorted bam (or sam) file which is nice and quick to view on IGV.
Best Wishes, David.
__________________________________ Dr David A. Matthews
Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K.
Tel. +44 117 3312058 Fax. +44 117 3312091
D.A.Matthews@bristol.ac.uk
On 22 Feb 2011, at 15:52, Ying Zhang wrote:
Dear David:
thank you very much for helping me!
I have download the IGV and I do find the IGVtools, however, I am not sure which tool I should use for generate a tdf file, the tile function will generate a tdf file, but the input file format does not include bam or sam file, instead it need wig file. But I have no wig file to put in. So I am wondering whether you need to use other tool first. I really appreciate your help! Thank you very much!
Best
Ying
Quoting David Matthews <D.A.Matthews@bristol.ac.uk>:
Hi,
You can get an equivalent visualisation from the IGV viewer by the Broad Institute - its under IGV tools and generates a tdf file from bam or sam files. This also gives a quick and easy way of looking at depth at any particular site and is very accessible.
Cheers David
On 21 Feb 2011, at 21:44, Jeremy Goecks wrote:
Hi all,
Ann is correct - Tophat does not produce .wig files when run anymore. However, it's fairly easy to use Galaxy to make a wiggle-like coverage file from a BAM file:
(a) run the pileup tool on your BAM to create a pileup file; (b) cut columns 1 and 4 to get your coverage file.
A final note: it's often difficult to visualize coverage files because they're so large. You might be better off visualizing the BAM file and using the coverage file for statistics.
Best, J.
Hello,
I think I know the answer (sort of) to this question.
This may be because newer versions of tophat stopped running the "wiggles" program, which is still part of the tophat distribution and is the program that makes the "coverage.wig" file.
A later version of tophat might bring this back, however - there's a note to this effect in the tophat python code.
So if you can run wiggles, you can make the "coverage.wig" file on your own.
A student here at UNC Charlotte (Adam Baxter) made a few changes to the "wiggles" source code that would allow you to use it with samtools to make a "coverage.wig" file from the "accepted_hits.bam" file that TopHat creates.
If you (or anyone else) would like a copy, please email Adam, who is cc'ed on this email.
We would be happy to help add it to Galaxy if this would be of interest to you or other Galaxy users.
If there is any way we can be of assistance, please let us know!
Very best wishes,
Ann Loraine
On 2/21/11 3:39 PM, "Ying Zhang" <ying.zhang.yz323@yale.edu> wrote:
Hi:
I am using tophat in galaxy to analyze my paired-end RNA-seq data and find out that after the tophat analysis, we can not get the wig file from it anymore which is used to be able to. Do you have any idea of how to still be able to get the wig file after tophat analysis? Thanks a lot!
Best
Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286 _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
-- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286
_______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list:
http://lists.bx.psu.edu/listinfo/galaxy-dev
To manage your subscriptions to this and other Galaxy lists, please use the interface at:
{kind=link}
{kind=link}

David, I have been also uisng the IGV like you have suggested. Vasu --- On Tue, 2/22/11, David Matthews <D.A.Matthews@bristol.ac.uk> wrote: From: David Matthews <D.A.Matthews@bristol.ac.uk> Subject: Re: [galaxy-user] get wig file after tophat To: "Ying Zhang" <ying.zhang.yz323@yale.edu> Cc: "Baxter, Adam" <Adam.Baxter@uncc.edu>, galaxy-user@bx.psu.edu Date: Tuesday, February 22, 2011, 10:54 AM HI, The option you need in IGV tools is "count". You set a window size and this gives you a tdf file from your sorted bam (or sam) file which is nice and quick to view on IGV. Best Wishes, David. __________________________________ Dr David A. Matthews Senior Lecturer in Virology Room E49 Department of Cellular and Molecular Medicine, School of Medical Sciences University Walk, University of Bristol Bristol. BS8 1TD U.K. Tel. +44 117 3312058 Fax. +44 117 3312091 D.A.Matthews@bristol.ac.uk On 22 Feb 2011, at 15:52, Ying Zhang wrote: Dear David: thank you very much for helping me! I have download the IGV and I do find the IGVtools, however, I am not sure which tool I should use for generate a tdf file, the tile function will generate a tdf file, but the input file format does not include bam or sam file, instead it need wig file. But I have no wig file to put in. So I am wondering whether you need to use other tool first. I really appreciate your help! Thank you very much! Best Ying Quoting David Matthews <D.A.Matthews@bristol.ac.uk>: Hi, You can get an equivalent visualisation from the IGV viewer by the Broad Institute - its under IGV tools and generates a tdf file from bam or sam files. This also gives a quick and easy way of looking at depth at any particular site and is very accessible. Cheers David On 21 Feb 2011, at 21:44, Jeremy Goecks wrote: Hi all, Ann is correct - Tophat does not produce .wig files when run anymore. However, it's fairly easy to use Galaxy to make a wiggle-like coverage file from a BAM file: (a) run the pileup tool on your BAM to create a pileup file; (b) cut columns 1 and 4 to get your coverage file. A final note: it's often difficult to visualize coverage files because they're so large. You might be better off visualizing the BAM file and using the coverage file for statistics. Best, J. Hello, I think I know the answer (sort of) to this question. This may be because newer versions of tophat stopped running the "wiggles" program, which is still part of the tophat distribution and is the program that makes the "coverage.wig" file. A later version of tophat might bring this back, however - there's a note to this effect in the tophat python code. So if you can run wiggles, you can make the "coverage.wig" file on your own. A student here at UNC Charlotte (Adam Baxter) made a few changes to the "wiggles" source code that would allow you to use it with samtools to make a "coverage.wig" file from the "accepted_hits.bam" file that TopHat creates. If you (or anyone else) would like a copy, please email Adam, who is cc'ed on this email. We would be happy to help add it to Galaxy if this would be of interest to you or other Galaxy users. If there is any way we can be of assistance, please let us know! Very best wishes, Ann Loraine On 2/21/11 3:39 PM, "Ying Zhang" <ying.zhang.yz323@yale.edu> wrote: Hi: I am using tophat in galaxy to analyze my paired-end RNA-seq data and find out that after the tophat analysis, we can not get the wig file from it anymore which is used to be able to. Do you have any idea of how to still be able to get the wig file after tophat analysis? Thanks a lot! Best Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286 _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ -- Ann Loraine Associate Professor Dept. of Bioinformatics and Genomics, UNCC North Carolina Research Campus 600 Laureate Way Kannapolis, NC 28081 704-250-5750 www.transvar.org _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/ Ying Zhang, M.D., Ph.D. Postdoctoral Associate Department of Genetics, Yale University School of Medicine 300 Cedar Street,S320 New Haven, CT 06519 Tel: (203)737-2616 Fax: (203)737-2286 -----Inline Attachment Follows----- _______________________________________________ The Galaxy User list should be used for the discussion of Galaxy analysis and other features on the public server at usegalaxy.org. For discussion of local Galaxy instances and the Galaxy source code, please use the Galaxy Development list: http://lists.bx.psu.edu/listinfo/galaxy-dev To manage your subscriptions to this and other Galaxy lists, please use the interface at: http://lists.bx.psu.edu/
participants (3)
-
 David Matthews
David Matthews -
 Jeremy Goecks
Jeremy Goecks -
 vasu punj
vasu punj